










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
ADENURIC 80 mg film-coated tablets
2.
Each tablet contains 80 mg of febuxostat.
Excipients: Each tablet contains 76.50 mg of lactose monohydrate
For a full list of excipients, see section 6.1.
3.
Film-coated tablet.
Pale yellow to yellow, film-coated, capsule shaped tablets, engraved with “80” on one side.
4.
Treatment of chronic hyperuricaemia in conditions where urate deposition has already occurred
(including a history, or presence of, tophus and/or gouty arthritis).
The recommended oral dose of ADENURIC is 80 mg once daily without regard to food. If serum uric
acid is > 6 mg/dl (357 µmol/l) after 2-4 weeks, ADENURIC 120 mg once daily may be considered.
ADENURIC works sufficiently quickly to allow retesting of the serum uric acid after 2 weeks. The
therapeutic target is to decrease and maintain serum uric acid below 6 mg/dl (357μmol/l).
Gout flare prophylaxis of at least 6 months is recommended (see section 4.4).
Special populations
Renal insufficiency
No dosage adjustment is necessary in patients with mild or moderate renal impairment. The efficacy
and safety have not been fully evaluated in patients with severe renal impairment (creatinine clearance
<30 ml/min, see section 5.2).
Hepatic impairment
The recommended dosage in patients with mild hepatic impairment is 80 mg. Limited information is
available in patients with moderate hepatic impairment.
The efficacy and safety of febuxostat has not
been studied in patients with severe hepatic impairment (Child Pugh Class C ).
Elderly
No dose adjustment is required in the elderly (see section 5.2).
Children and adolescents
As there has been no experience in children and adolescents, the use of febuxostat in such patients is
not recommended.
2
Organ transplant recipients
As there has been no experience in organ transplant recipients, the use of febuxostat in such patients is
not recommended (see section 5.1).
Hypersensitivity to the active substance or to any of the excipients (see section 4.8).
Cardio-vascular disorders
Treatment with febuxostat in patients with ischaemic heart disease or congestive heart failure is not
recommended (see section 4.8).
Acute gouty attacks (gout flare)
Febuxostat treatment should not be started until an acute attack of gout has completely subsided. As
with other urate lowering medicinal products, gout flares may occur during initiation of treatment due
to changing serum uric acid levels resulting in mobilization of urate from tissue deposits (see section
4.8 and 5.1). At treatment initiation with febuxostat flare prophylaxis for at least 6 months with an
NSAID or colchicine is recommended (see section 4.2).
If a gout flare occurs during febuxostat treatment, it should not be discontinued. The gout flare should
be managed concurrently as appropriate for the individual patient. Continuous treatment with
febuxostat decreases frequency and intensity of gout flares.
Xanthine deposition
As with other urate lowering medicinal products, in patients in whom the rate of urate formation is
greatly increased (e.g. malignant disease and its treatment, Lesch-Nyhan syndrome) the absolute
concentration of xanthine in urine could, in rare cases, rise sufficiently to allow deposition in the
urinary tract. As there has been no experience with febuxostat, its use in these populations is not
recommended.
Mercaptopurine/azathioprine
Febuxostat use is not recommended in patients concomitantly treated with
mercaptopurine/azathioprine (see section 4.5).
Theophylline
Febuxostat should be used with caution in patients concomitantly treated with theophylline and
theophylline levels should be monitored in patients starting febuxostat therapy (see section 4.5).
Liver disorders
During the combined phase 3 clinical studies, mild liver function test abnormalities were observed in
patients treated with febuxostat (5.0%). Liver function test is recommended prior to the initiation of
therapy with febuxostat and periodically thereafter based on clinical judgment (see section 5.1).
Thyroid disorders
Increased TSH values (>5.5 µIU/ml) were observed in patients on long-term treatment with febuxostat
(5.5%) in the long term open label extension studies. Caution is required when febuxostat is used in
patients with alteration of thyroid function (see section 5.1).
Lactose
Febuxostat tablets contain lactose. Patients with rare hereditary problems of galactose intolerance, the
Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
3
Mercaptopurine/azathioprine
Although interaction studies with febuxostat have not been performed, inhibition of xanthine oxidase
(XO) is known to result in an increase in mercaptopurine or azathioprine levels. On the basis of the
mechanism of action of febuxostat on XO inhibition concomitant use is not recommended.
Drug interaction studies of febuxostat with cytotoxic chemotherapy have not been conducted. No data
is available regarding the safety of febuxostat during cytotoxic therapy.
Theophylline
Although interaction studies have not been performed with febuxostat, inhibition of XO may cause an
increase in the theophylline level (inhibition of the metabolism of theophylline has been reported with
other XO inhibitors). Hence caution is advised if these active substances are given concomitantly, and
theophylline levels should be monitored in patients starting febuxostat therapy.
Naproxen and other inhibitors of glucuronidation
Febuxostat metabolism depends on UGT enzymes. Medicinal products that inhibit glucuronidation,
such as NSAIDs and probenecid, could in theory affect the elimination of febuxostat. In healthy
subjects concomitant use of febuxostat and naproxen 250mg BID was associated with an increase in
febuxostat exposure (C
max
28%, AUC 41% and t
1/2
26%). In clinical studies the use of naproxen or
other NSAIDs/Cox-2 inhibitors was not related to any clinically significant increase in adverse events.
Febuxostat can be co-administered with naproxen with no dose adjustment of febuxostat or naproxen
being necessary.
Inducers of glucuronidation
Potent inducers of UGT enzymes might possibly lead to increased metabolism and decreased efficacy
of febuxostat. Monitoring of serum uric acid is therefore recommended 1-2 weeks after start of
treatment with a potent inducer of glucuronidation. Conversely, cessation of treatment of an inducer
might lead to increased plasma levels of febuxostat.
Colchicine/indometacin/hydrochlorothiazide/warfarin
Febuxostat can be co-administered with colchicine or indomethacin with no dose adjustment of
febuxostat or the co-administered active substance being necessary.
No dose adjustment is necessary for febuxostat when administered with hydrochlorothiazide.
No dose adjustment is necessary for warfarin when administered with febuxostat. Administration of
febuxostat (80 mg or 120 mg once daily) with warfarin had no effect on the pharmacokinetics of
warfarin in healthy subjects. INR and Factor VII activity were also not affected by the co-
administration of febuxostat.
Desipramine/CYP2D6 substrates.
Febuxostat was shown to be a weak inhibitor of CYP2D6
in vitro.
In a study in healthy subjects,
120 mg ADENURIC QD resulted in a mean 22% increase in AUC of desipramine, a CYP2D6
substrate indicating a potential weak inhibitory effect of febuxostat on the CYP2D6 enzyme
in vivo
.
Thus, co-administration of febuxostat with other CYP2D6 substrates is not expected to require any
dose adjustment for those compounds.
Antacids
Concomitant ingestion of an antacid containing magnesium hydroxide and aluminium hydroxide has
been shown to delay absorption of febuxostat (approximately 1 hour) and to cause a 32% decrease in
C
max
, but no significant change in AUC was observed. Therefore, febuxostat may be taken without
regard to antacid use.
4
Pregnancy
Data on a very limited number of exposed pregnancies have not indicated any adverse effects of
febuxostat on pregnancy or on the health of the foetus/new born child. Animal studies do not indicate
direct or indirect harmful effects with respect to pregnancy,
embryonal/foetal development or
parturition (see section 5.3).
The potential risk for human is unknown. Febuxostat should not be used
during pregnancy.
Breast feeding
It is unknown whether febuxostat is excreted in human breast milk. Animal studies have shown
excretion of this active substance in breast milk and an impaired development of suckling pups. A risk
to a suckling infant cannot be excluded. Febuxostat should not be used while breast-feeding.
No studies on the effects on the ability to drive and use machines have been performed. As with other
xanthine oxidase inhibitors adverse reactions such as somnolence, dizziness and paraesthesia have
been reported. Patients should exercise caution before driving, using machinery or participating in
dangerous activities until they are reasonably certain that ADENURIC does not adversely affect
performance.
A total of 4,072 subjects received at least one dose of ADENURIC (10 mg – 300 mg) in clinical
studies.
Combined phase 3 randomised controlled studies
In randomised controlled phase 3 clinical studies, >2,500 patients have been treated with 40 mg to
120 mg (1513 subjects enrolled in a 26-week study (CONFIRMS), 536 subjects enrolled in a 28-week
study (APEX) and 507 subjects enrolled in a 52-weeks study (FACT)). The treatment-related events
(ADRs) were mostly mild or moderate in severity.
The most commonly reported ADRs (investigator assessment) are liver function abnormalities (5.0%),
diarrhoea (2.7%), nausea (1.3%), headache (1.2%), rash (1.2 %).
Gout flares were also commonly observed soon after the start of treatment and during the first months.
Thereafter, the frequency of gout flare decreases in a time-dependent manner. As with other urate
lowering medicinal products, gout flare prophylaxis is recommended (see section 4.2 and 4.4).
A numerical greater incidence of investigator-reported cardiovascular APTC events (defined endpoints
from the Anti-Platelet Trialists’ Collaboration (APTC) including cardiovascular death, non-fatal
myocardial infarction, non-fatal stroke) was observed in the febuxostat total group compared to the
allopurinol group in the APEX and FACT studies (1.3 vs. 0.3 events per 100 PYs), but not in the
CONFIRMS study. The incidence of investigator-reported cardiovascular APTC events in the
combined Phase III studies (APEX, FACT and CONFIRMS studies) was 0.7 vs. 0.6 events per 100
PYs. In the long-term extension studies the incidences of investigator-reported APTC events were 1.2
and 0.6 events per 100 PYs for febuxostat and allopurinol, respectively. No statistically significant
differences were found and no causal relationship with febuxostat was established. Identified risk
factors among these patients were a medical history of atherosclerotic disease and/or myocardial
infarction, or of congestive heart failure.
Common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100) and rare (≥ 1/10,000 to < 1/1000)
adverse reactions suspected (investigator assessment) to be drug related occurring in patients treated
with 40 mg to 120 mg febuxostat and reported more than once in the total febuxostat treatment group
are listed below.
5
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Table 1: Treatment related adverse reactions in combined phase 3 randomized controlled studies
Blood and lymphatic system
disorders
Rare
Pancytopenia
Metabolism and nutrition
disorders
Common***
Gout flares
Uncommon
Decrease appetite
Rare
Weight increase/decrease, increase appetite, anorexia,
hyperlipidemia
Psychiatric disorders
Uncommon
Libido decreased, insomnia
Rare
Nervousness
Nervous system disorders
Common
Headache
Uncommon
Dizziness, paraesthesia, somnolence, altered taste, hypoaesthesia
Ear and labyrinth disorders
Rare
Tinnitus
Cardiac disorders
Uncommon
Atrial fibrillation, palpitations, ECG abnormal
Vascular disorders
Uncommon
Hypertension, flushing, hot flush
Respiratory system disorders
Uncommon
Dyspnoea, upper respiratory tract infection
Gastrointestinal disorders
Common
Diarrhoea
*
, nausea
Uncommon:
Abdominal pain, gastro-oesophageal reflux disease, vomiting, dry
mouth, dyspepsia, constipation, frequent
stools, flatulence,
gastrointestinal discomfort
Rare
Pancreatitis, mouth ulceration
Hepato-biliary disorders
Common
Liver function abnormalities*
Skin and subcutaneous tissue
disorders
Common
Rash
**
Uncommon
Dermatitis, urticaria, pruritus
Rare
Alopecia, hyperhidrosis
Musculoskeletal and connective
tissue disorders
Uncommon
Arthralgia, myalgia, musculoskeletal pain, muscle weakness,
muscle spasm
6














Rare
Arthritis, joint stiffness, musculoskeletal stiffness
Renal and urinary disorders
Uncommon
Nephrolithiasis, haematuria, pollakiuria, renal failure
Rare
Micturition urgency
Reproductive system and breast
disorder
Rare
Erectile dysfunction
General disorders and
administration site conditions
Uncommon
Fatigue, oedema, chest pain, chest discomfort
Rare
Thirst
Investigations
Uncommon
Blood amylase increase, platelet count decrease, blood creatinine
increase, haemoglobin decrease, blood urea increase, blood
triglycerides increase, blood cholesterol increase, haematocritic
decrease, blood lactate dehydrogenase increased
Rare
Blood glucose increase, activated partial thromboplastin time
prolonged, red blood cell count decrease, blood alkaline
phosphatase increase
*
Treatment-emergent non-infective diarrhoea and abnormal liver function tests in the combined
Phase III studies are more frequent in patients concomitantly treated with colchicine.
**
No serious rashes or severe hypersensitivity reactions were noted in the clinical studies.
*** See section 5.1 for incidences of gout flares in the individual Phase 3 randomized
controlled studies.
Long-term open label extension studies
In the long-term open label extension studies (1143 patients), the number of patients treated with
febuxostat 40 mg/80 mg/120 mg up to 1 year was 909, up to 2 years was 781, up to 3 years was 348,
and up to 4 and 5 years was 60. The treatment-related events reported during the long-term extension
studies were similar to those reported in the Phase 3 studies (see Table 1). The most commonly
reported treatment-related events (investigator assessment) are: liver function abnormalities, diarrhoea,
headache, rash, hypertension, oedema.
The following treatment-related events were reported more than once in the total febuxostat treatment
group and were reported as uncommon in patients taking febuxostat 40 mg/80 mg/120 mg in long-
term extension studies (up to 5 years, >2,660 Patient-years of exposure). These treatment-related
events were either not reported or reported at a lower frequency for these doses, in the combined Phase
3 studies: abdominal distension, cholelithiasis, bronchitis, weight increase, blood creatine increase,
diabetes mellitus, hyperlipidaemia, arthritis, muscle tightness, hyposmia, hemiparesis, cough, skin
discolouration, skin lesion, bursitis, proteinuria, petechiae, erectile dysfunction, blood potassium
increase, blood TSH increase, lymphocyte count decreased, WBC decrease.
No case of overdose has been reported. Patients with an overdose should be managed by symptomatic
and supportive care.
7








5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group:
Preparations inhibiting uric acid production,
ATC code: M04AA03
Mechanism of action
Uric acid is the end product of purine metabolism in humans and is generated in the cascade of
hypoxanthine → xanthine → uric acid. Both steps in the above transformations are catalyzed by
xanthine oxidase (XO). Febuxostat is a 2-arylthiazole derivative that achieves its therapeutic effect of
decreasing serum uric acid by selectively inhibiting XO. Febuxostat is a potent, non-purine selective
inhibitor of XO (NP-SIXO) with an
in vitro
inhibition Ki value less than one nanomolar. Febuxostat
has been shown to potently inhibit both the oxidized and reduced forms of XO. At therapeutic
concentrations febuxostat does not inhibit other enzymes involved in purine or pyrimidine
metabolism, namely, guanine deaminase, hypoxanthine guanine phosphoribosyltransferase, orotate
phosphoribosyltransferase, orotidine monophosphate decarboxylase or purine nucleoside
phosphorylase.
Clinical studies results
The efficacy of ADENURIC was demonstrated in three Phase 3 pivotal studies (the two pivotal APEX
and FACT studies, and the additional CONFIRMS study described below) that were conducted in
4101 patients with hyperuricemia and gout. In each phase 3 pivotal study, ADENURIC demonstrated
superior ability to lower and maintain serum uric acid levels compared to allopurinol. The primary
efficacy endpoint in the APEX and FACT studies was the proportion of patients whose last 3 monthly
serum uric acid levels were < 6.0 mg/dl (357 µmol/l). In the additional phase 3 CONFIRMS study, for
which results became available after the marketing authorisation for ADENURIC was first issued, the
primary efficacy endpoint was the proportion of patients whose serum urate level was < 6.0 mg/dL at
the final visit. No patients with organ transplant have been included in these studies (see section 4.2).
APEX Study
: The Allopurinol and Placebo-Controlled Efficacy Study of Febuxostat (APEX) was a
Phase 3, randomized, double-blind, multicenter, 28-week study. One thousand and seventy-two (1072)
patients were randomized: placebo (n=134), ADENURIC 80 mg QD (n=267), ADENURIC 120 mg
QD (n=269), ADENURIC 240 mg QD (n=134) or allopurinol (300 mg QD [n=258] for patients with a
baseline serum creatinine ≤1.5 mg/dl or 100 mg QD [n=10] for patients with a baseline serum
creatinine >1.5 mg/dl and ≤2.0 mg/dl). Two hundred and forty mg febuxostat (2 times the
recommended highest dose) was used as a safety evaluation dose.
The APEX study showed statistically significant superiority of both the ADENURIC 80 mg QD and
the ADENURIC 120 mg QD treatment arms
versus
the conventionally used doses of allopurinol
300mg (n = 258) /100mg (n = 10) treatment arm in reducing the sUA below 6 mg/dl (357 µmol/l)
(see Table 2 and Figure 1).
FACT Study
: The Febuxostat Allopurinol Controlled Trial (FACT) Study was a Phase 3, randomized,
double-blind, multicenter, 52-week study. Seven hundred sixty (760) patients were randomized:
ADENURIC 80 mg QD (n=256), ADENURIC 120 mg QD (n=251), or allopurinol 300 mg QD
(n=253).
The FACT study showed the statistically significant superiority of both ADENURIC 80 mg and
ADENURIC 120 mg QD treatment arms
versus
the conventionally used dose of allopurinol 300 mg
treatment arm in reducing and maintaining sUA below 6 mg/dl (357 µmol/l).
8
Table 2 summarises the primary efficacy endpoint results:
Table 2
Proportion of Patients with Serum Uric Acid Levels <6.0 mg/dl (357µmol/l)
Last Three Monthly Visits
Study
ADENURIC
80 mg QD
ADENURIC
120 mg QD
Allopurinol
300 /
100 mg QD
1
APEX
(28 weeks)
48%
*
(n=262)
65%
*, #
(n=269)
22%
(n=268)
FACT
(52 weeks)
53%
*
(n=255)
62%
*
(n=250)
21%
(n=251)
22%
(n=519)
1
results from subjects receiving either 100 mg QD (n=10:
patients with
serum creatinine >1.5 and ≤2.0 mg/dl) or 300 mg QD (n=509) were pooled
for analyses.
* p < 0.001 vs allopurinol,
#
p < 0.001 vs 80 mg
51%
*
(n=517)
63%
*, #
(n=519)
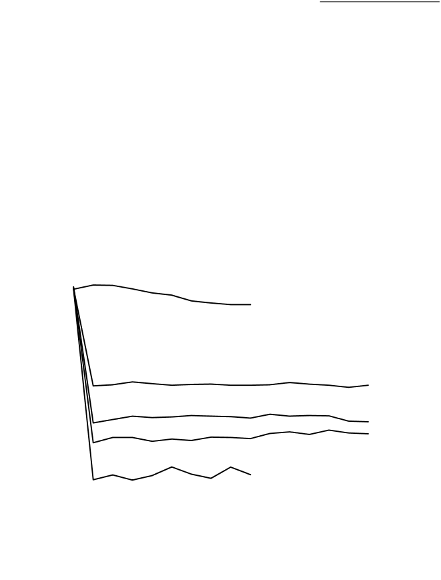
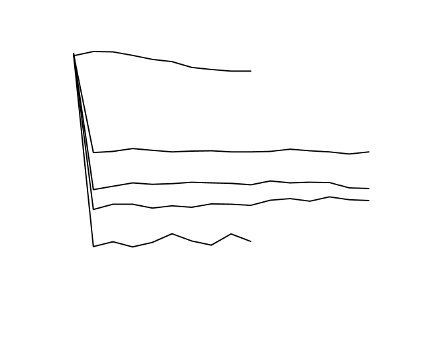
The ability of ADENURIC to lower serum uric acid levels was prompt and persistent. Reduction in
serum uric acid level to <6.0 mg/dl (357 µmol/l) was noted by the Week 2 visit and was maintained
throughout treatment. The mean serum uric acid levels over time for each treatment group from the
two pivotal Phase 3 studies are shown in Figure 1.
Figure 1 Mean Serum Uric Acid Levels in Combined Pivotal Phase 3 Studies
11
10
Placebo
9
8
7
Allopurinol
6
ADE
NU
RI
C
80
mg
5
4
ADENURIC 120 mg
3
2
ADENURIC 240 mg
BL 2 4 6 8 12 16 20 24 28 32 36 40 44 48 52
Week
BL=baseline SEM=standard error of the mean
Note:
509 patients received allopurinol 300 mg QD; 10 patients with serum creatinine >1.5 and < 2.0
mg/dl were dosed with 100 mg QD. (10 patients out of 268 in APEX study).
240 mg febuxostat was used to evaluate the safety of febuxostat at twice the recommended
highest dose.
9
Combined
Results











































































































































































































































CONFIRMS Study: The CONFIRMS study was a Phase 3, randomized, controlled, 26-week study to
evaluate the safety and efficacy of febuxostat 40 mg and 80 mg, in comparison with allopurinol 300
mg or 200 mg, in patients with gout and hyperuricaemia. Two thousand and two hundred-sixty nine
(2269) patients were randomized: ADENURIC 40 mg QD (n=757), ADENURIC 80 mg QD (n=756),
or allopurinol 300/200 mg QD (n=756). At least 65% of the patients had mild-moderate renal
impairment (with creatinine clearance of 30-89 mL/min). Prophylaxis against gout flares was
obligatory over the 26-week period.
The proportion of patients with serum urate levels of < 6.0 mg/dL (357 µmol/L) at the final visit, was
45% for 40 mg febuxostat, 67% for febuxostat 80 mg and 42% for
allopurinol 300/200 mg,
respectively.
Primary endpoint in the sub-group of patients with renal impairment
The APEX Study evaluated efficacy in 40 patients with renal impairment (i.e., baseline serum
creatinine > 1.5 mg/dl and ≤2.0 mg/dl). For renally impaired subjects who were randomized to
allopurinol, the dose was capped at 100mg QD. ADENURIC achieved the primary efficacy endpoint
in 44% (80 mgQD), 45% (120 mg QD), and 60% (240 mg QD) of patients compared to 0% in the
allopurinol 100 mg QD and placebo groups.
There were no clinically significant differences in the percent decrease in serum uric acid
concentration in healthy subjects irrespective of their renal function (58 % in the normal renal function
group and 55% in the severe renal dysfunction group).
An analysis in patients with gout and renal impairment was prospectively defined in the CONFIRMS
study, and showed that febuxostat was significantly more efficacious in lowering serum urate levels to
< 6 mg/dL compared to allopurinol 300 mg/200 mg in patients who had gout with mild to moderate
renal impairment (65% of patients studied).
Primary endpoint in the sub group of patients with sUA ≥ 10 mg/dl
Approximately 40% of patients (combined APEX and FACT) had a baseline sUA of ≥ 10 mg/dl. In
this subgroup ADENURIC achieved the primary efficacy endpoint (sUA < 6.0 mg/dL at the last 3
visits) in 41% (80 mg QD), 48% (120 mg QD), and 66% (240 mg QD) of patients compared to 9% in
the allopurinol 300 mg/100 mg QD and 0 % in the placebo groups.
In the CONFIRMS study, the proportion of patients achieving the primary efficacy endpoint (sUA <
6.0 mg/dL at the final visit) for patients with a baseline serum urate level of ≥ 10 mg/dL treated with
febuxostat 40 mg QD was 27% (66/249), with febuxostat 80 mg QD 49% (125/254) and with
allopurinol 300 mg/200 mg QD 31% (72/230), respectively.
Clinical Outcomes: proportion of patients requiring treatment for a gout flare
APEX study: During the 8-week prophylaxis period, a greater proportion of subjects in the febuxostat
120 mg (36%) treatment group required treatment for gout flare compared to febuxostat 80 mg (28%),
allopurinol 300 mg (23%) and placebo (20%). Flares increased following the prophylaxis period and
gradually decreased over time. Between 46% and 55% of subjects received treatment for gout flares
from Week 8 and Week 28. Gout flares during the last 4 weeks of the study (Weeks 24-28) were
observed in 15% (febuxostat 80, 120 mg), 14% (allopurinol 300 mg) and 20% (placebo) of subjects.
FACT study: During the 8-week prophylaxis period, a greater proportion of subjects in the febuxostat
120 mg (36%) treatment group required treatment for a gout flare compared to both the febuxostat 80
mg (22%) and allopurinol 300 mg (21%) treatment groups. After the 8-week prophylaxis period, the
incidences of flares increased and gradually decreased over time (64% and 70% of subjects received
treatment for gout flares from Week 8-52). Gout flares during the last 4 weeks of the study (Weeks 49-
52) were observed in 6-8% (febuxostat 80 mg, 120 mg) and 11% (allopurinol 300 mg) of subjects.
The proportion of subjects requiring treatment for a gout flare (APEX and FACT Study) was
numerically lower in the groups that achieved an average post-baseline serum urate level <6.0 mg/dl,
<5.0 mg/dl, or <4.0 mg/dl compared to the group that achieved an average post-baseline serum urate
10
level ≥6.0 mg/dl during the last 32 weeks of the treatment period (Week 20-Week 24 to Week 49 - 52
intervals).
During the CONFIRMS study, the percentages of patients who required treatment for gout flares (Day
1 through Month 6) were 31% and 25% for the febuxostat 80 mg and allopurinol groups, respectively.
No difference in the proportion of patients requiring treatment for gout flares was observed between
the febuxostat 80 mg and 40 mg groups.
Long-term, open label extension Studies
EXCEL Study (C02-021): The Excel study was a three years Phase III, open label, multicenter,
randomised, allopurinol-controlled, safety extension study for patients who had completed the pivotal
Phase III studies (APEX or FACT). A total of 1086 patients were enrolled: ADENURIC 80 mg QD
(n=649), Adenuric 120 mg QD (n=292) and allopurinol 300/100 mg QD (n=145). About 69 % of
patients required no treatment change to achieve a final stable treatment. Patients who had 3
consecutive sUA levels >6.0 mg/dl were withdrawn.
Serum urate levels were maintained over time (i.e. 91% and 93% of patients on initial treatment with
febuxostat 80 mg and 120 mg, respectively, had sUA <6 mg/dl at Month 36).
Three years data showed a decrease in the incidence of gout flares with less than 4 % of patients
requiring treatment for a flare (i.e. more than 96 % of patients did not require treatment for a flare) at
Month 16-24 and at Month 30-36.
46% and 38%, of patients on final stable treatment of febuxostat 80 or 120 mg QD, respectively, had
complete resolution of the primary palpable tophus from baseline to the Final Visit.
FOCUS Study (TMX-01-005) was a 5 years Phase II, open-label, multicenter, safety extension study
for patients who had completed the febuxostat 4 weeks of double blind dosing in study TMX-00-004.
116 patients were enrolled and received initially febuxostat 80 mg QD. 62 % of patients required no
dose adjustment to maintain sUA <6 mg/dL and 38 % of patients required a do se adjustment to
achieve a final stable dose.
The proportion of patients with serum urate levels of <6.0 mg/dL (357 µmol/l) at the final visit was
greater than 80% (81-100%) at each febuxostat dose.
During the phase 3 clinical studies, mild liver function test abnormalities were observed in patients
treated with febuxostat (5.0%). These rates were similar to the rates reported on allopurinol (4.2%)
(see section 4.4). Increased TSH values (>5.5 µIU/ml) were observed in patients on long-term
treatment with febuxostat (5.5%) and patients with allopurinol (5.8%) in the long term open label
extension studies (see section 4.4).
5.2 Pharmacokinetic properties
In healthy subjects, maximum plasma concentrations (C
max
) and area under the plasma concentration
time curve (AUC) of febuxostat increased in a dose proportional manner following single and multiple
doses of 10 mg to 120 mg. For doses between 120 mg and 300 mg, a greater than dose proportional
increase in AUC is observed for febuxostat. There is no appreciable accumulation when doses of
10 mg to 240 mg are administered every 24 hours. Febuxostat has an apparent mean terminal
elimination half-life (t
1/2
) of approximately 5 to 8 hours.
Population pharmacokinetic/pharmacodynamic analyses were conducted in 211 patients with
hyperuricemia and gout, treated with ADENURIC 40-240 mg QD. In general, febuxostat
pharmacokinetic parameters estimated by these analyses are consistent with those obtained from
healthy subjects, indicating that healthy subjects are representative for
pharmacokinetic/pharmacodynamic assessment in the patient population with gout.
11
Absorption
Febuxostat is rapidly (t
max
of 1.0-1.5 h) and well absorbed (at least 84%). After single or multiple oral
80 and 120 mg once daily doses, C
max
is approximately 2.8-3.2 µg/ml, and 5.0-5.3 µg/ml, respectively.
Absolute bioavailability of the febuxostat tablet formulation has not been studied.
Following multiple oral 80 mg once daily doses or a single 120 mg dose with a high fat meal, there
was a 49% and 38% decrease in C
max
and a 18% and 16% decrease in AUC, respectively. However, no
clinically significant change in the percent decrease in serum uric acid concentration was observed
where tested (80 mg multiple dose). Thus, ADENURIC may be taken without regard to food.
Distribution
The apparent steady state volume of distribution (V
ss
/F) of febuxostat ranges from 29 to 75 l after oral
doses of 10-300 mg. The plasma protein binding of febuxostat is approximately 99.2%, (primarily to
albumin), and is constant over the concentration range achieved with 80 and 120 mg doses. Plasma
protein binding of the active metabolites ranges from about 82% to 91%.
Metabolism
Febuxostat is extensively metabolized by conjugation
via
uridine diphosphate glucuronosyltransferase
(UDPGT) enzyme system and oxidation
via
the cytochrome P450 (CYP) system. Four
pharmacologically active hydroxyl metabolites have been identified, of which three occur in plasma of
humans.
In vitro
studies with human liver microsomes showed that those oxidative metabolites were
formed primarily by CYP1A1, CYP1A2,
CYP2C8 or CYP2C9 and febuxostat glucuronide was formed mainly by UGT 1A1, 1A8, and 1A9
Elimination
Febuxostat is eliminated by both hepatic and renal pathways. Following an 80 mg oral dose of
14
C-
labeled febuxostat, approximately 49% of the dose was recovered in the urine as unchanged febuxostat
(3%), the acyl glucuronide of the active substance (30%), its known oxidative metabolites and their
conjugates (13%), and other unknown metabolites (3%). In addition to the urinary excretion,
approximately 45% of the dose was recovered in the faeces as the unchanged febuxostat (12%), the
acyl glucuronide of the active substance (1%), its known oxidative metabolites and their conjugates
(25%), and other unknown metabolites (7%).
Special populations
Renal insufficiency
Following multiple doses of 80 mg of ADENURIC in patients with mild, moderate or severe renal
insufficiency, the C
max
of febuxostat did not change, relative to subjects with normal renal function.
The mean total AUC of febuxostat increased by approximately 1.8-fold from 7.5 μg⋅h/ml in the
normal renal function group to 13.2 μg.h/ml in the severe renal dysfunction group. The C
max
and AUC
of active metabolites increased up to 2- and 4-fold, respectively. However, no dose adjustment is
necessary in patients with mild or moderate renal impairment.
Hepatic impairment
Following multiple doses of 80 mg of ADENURIC in patients with mild (Child-Pugh Class A) or
moderate (Child-Pugh Class B) hepatic impairment, the C
max
and AUC of febuxostat and its
metabolites did not change significantly compared to subjects with normal hepatic function. No
studies have been conducted in patients with severe hepatic impairment (Child-Pugh Class C).
Age
There were no significant changes observed in AUC of febuxostat or its metabolites following
multiple oral doses of ADENURIC in elderly as compared to younger healthy subjects.
Gender
Following multiple oral doses of ADENURIC, the C
max
and AUC were 24% and 12% higher in
females than in males, respectively. However, weight-corrected C
max
and AUC were similar between
the genders. No dose adjustment is needed based on gender.
12
5.3 Preclinical safety data
Effects in non-clinical studies were generally observed at exposures in excess of the maximum human
exposure.
Carcinogenesis, mutagenesis, impairment of fertility
In male rats, a statistically significant increase in urinary bladder tumours (transitional cell papilloma
and carcinoma) was found only in association with xanthine calculi in the high dose group, at
approximately 11 times human exposure. There was no significant increase in any other tumour type
in either male or female mice or rats. These findings are considered a consequence of species specific
purine metabolism and urine composition and of no relevance to clinical use.
A standard battery of test for genotoxicity did not reveal any biologically relevant genotoxic effects
for febuxostat.
Febuxostat at oral doses up to 48 mg/kg/day was found to have no effect on fertility and reproductive
performance of male and female rats.
There was no evidence of impaired fertility, teratogenic effects, or harm to the foetus due to
febuxostat. There was high dose maternal toxicity accompanied by a reduction in weaning index and
reduced development of offspring in rats at approximately 4.3 times human exposure. Teratology
studies, performed in pregnant rats at approximately 4.3 times and pregnant rabbits at approximately
13 times human exposure did not reveal any teratogenic effects.
6.
6.1 List of excipients
Tablet core
Lactose monohydrate
Microcrystalline cellulose
Magnesium stearate
Hydroxypropylcellulose
Croscarmellose sodium
Silica, colloidal hydrated
Tablet coating
Opadry II, Yellow, 85F42129 containing:
Polyvinyl alcohol
Titanium dioxide (E171)
Macrogols 3350
Talc
Iron oxide yellow (E172)
6.2 Incompatibilities
Not applicable.
6.3 Shelf life
3 years
6.4 Special precautions for storage
This medicinal product does not require any special storage condition.
13
6.5 Nature and contents of container
Clear (Aclar/PVC/Aluminium) blister of 14 tablets.
ADENURIC 80 mg is available in pack sizes of 14, 28, 42, 56, 84 and 98 film-coated tablets.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal
No special requirements.
7.
Menarini International Operations Luxembourg S.A.
1, Avenue de la Gare, L-1611 Luxembourg
Luxembourg
8.
EU/1/08/447/001
EU/1/08/447/002
EU/1/08/447/005
EU/1/08/447/006
EU/1/08/447/007
EU/1/08/447/008
9.
21/04/2008
Detailed information on this product is available on the website of the European Medicines Agency
14
1.
ADENURIC 120 mg film-coated tablets
2.
Each tablet contains 120 mg of febuxostat.
Excipients: Each tablet contains 114.75mg of lactose monohydrate
For a full list of excipients, see section 6.1.
3.
Film-coated tablet.
Pale yellow to yellow, film-coated, capsule shaped tablets, engraved with “120” on one side
4.
Treatment of chronic hyperuricaemia in conditions where urate deposition has already occurred
(including a history, or presence of, tophus and/or gouty arthritis).
The recommended oral dose of ADENURIC is 80 mg once daily without regard to food. If serum uric
acid is > 6 mg/dl (357 µmol/l) after 2-4 weeks, ADENURIC 120 mg once daily may be considered.
ADENURIC works sufficiently quickly to allow retesting of the serum uric acid after 2 weeks. The
therapeutic target is to decrease and maintain serum uric acid below 6 mg/dl (357μmol/l).
Gout flare prophylaxis of at least 6 months is recommended (see section 4.4).
Special populations
Renal insufficiency
No dosage adjustment is necessary in patients with mild or moderate renal impairment. The efficacy
and safety have not been fully evaluated in patients with severe renal impairment (creatinine clearance
<30 ml/min, see section 5.2).
Hepatic impairment
The recommended dosage in patients with mild hepatic impairment is 80 mg. Limited information is
available in patients with moderate hepatic impairment.
The efficacy and safety of febuxostat has not
been studied in patients with severe hepatic impairment (Child Pugh Class C ).
Elderly
No dose adjustment is required in the elderly (see section 5.2).
Children and adolescents
As there has been no experience in children and adolescents, the use of febuxostat in such patients is
not recommended.
15
Organ transplant recipients
As there has been no experience in organ transplant recipients, the use of febuxostat in such patients is
not recommended (see section 5.1).
Hypersensitivity to the active substance or to any of the excipients (see section 4.8).
Cardio-vascular disorders
Treatment with febuxostat in patients with ischaemic heart disease or congestive heart failure is not
recommended (see section 4.8).
Acute gouty attacks (gout flare)
Febuxostat treatment should not be started until an acute attack of gout has completely subsided. As
with other urate lowering medicinal products, gout flares may occur during initiation of treatment due
to changing serum uric acid levels resulting in mobilization of urate from tissue deposits (see sections
4.8 and 5.1). At treatment initiation with febuxostat flare prophylaxis for at least 6 months with an
NSAID or colchicine is recommended (see section 4.2).
If a gout flare occurs during febuxostat treatment, it should not be discontinued. The gout flare should
be managed concurrently as appropriate for the individual patient. Continuous treatment with
febuxostat decreases frequency and intensity of gout flares.
Xanthine deposition
As with other urate lowering medicinal products, in patients in whom the rate of urate formation is
greatly increased (e.g. malignant disease and its treatment, Lesch-Nyhan syndrome) the absolute
concentration of xanthine in urine could, in rare cases, rise sufficiently to allow deposition in the
urinary tract. As there has been no experience with febuxostat, its use in these populations is not
recommended.
Mercaptopurine/azathioprine
Febuxostat use is not recommended in patients concomitantly treated with
mercaptopurine/azathioprine (see section 4.5).
Theophylline
Febuxostat should be used with caution in patients concomitantly treated with theophylline and
theophylline levels should be monitored in patients starting febuxostat therapy (see section 4.5).
Liver disorders
During the combined phase 3 clinical studies, mild liver function test abnormalities were observed in
patients treated with febuxostat (5.0%). Liver function test is recommended prior to the initiation of
therapy with febuxostat and periodically thereafter based on clinical judgement (see section 5.1).
Thyroid disorders
Increased TSH values (>5.5 µIU/ml) were observed in patients on long-term treatment with febuxostat
(5.5%) in the long term open label extension studies. Caution is required when febuxostat is used in
patients with alteration of thyroid function (see section 5.1).
Lactose
Febuxostat tablets contain lactose. Patients with rare hereditary problems of galactose intolerance, the
Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
16
Mercaptopurine/azathioprine
Although interaction studies with febuxostat have not been performed, inhibition of xanthine oxidase
(XO) is known to result in an increase in mercaptopurine or azathioprine levels. On the basis of the
mechanism of action of febuxostat on XO inhibition concomitant use is not recommended.
Drug interaction studies of febuxostat with cytotoxic chemotherapy have not been conducted. No data
is available regarding the safety of febuxostat during cytotoxic therapy.
Theophylline
Although interaction studies have not been performed with febuxostat, inhibition of XO may cause an
increase in the theophylline level (inhibition of the metabolism of theophylline has been reported with
other XO inhibitors). Hence caution is advised if these active substances are given concomitantly, and
theophylline levels should be monitored in patients starting febuxostat therapy.
Naproxen and other inhibitors of glucuronidation
Febuxostat metabolism depends on UGT enzymes. Medicinal products that inhibit glucuronidation,
such as NSAIDs and probenecid, could in theory affect the elimination of febuxostat. In healthy
subjects concomitant use of febuxostat and naproxen 250mg BID was associated with an increase in
febuxostat exposure (C
max
28%, AUC 41% and t
1/2
26%). In clinical studies the use of naproxen or
other NSAIDs/Cox-2 inhibitors was not related to any clinically significant increase in adverse events.
Febuxostat can be co-administered with naproxen with no dose adjustment of febuxostat or naproxen
being necessary.
Inducers of glucuronidation
Potent inducers of UGT enzymes might possibly lead to increased metabolism and decreased efficacy
of febuxostat. Monitoring of serum uric acid is therefore recommended 1-2 weeks after start of
treatment with a potent inducer of glucuronidation. Conversely, cessation of treatment of an inducer
might lead to increased plasma levels of febuxostat.
Colchicine/indometacin/hydrochlorothiazide/warfarin
Febuxostat can be co-administered with colchicine or indomethacin with no dose adjustment of
febuxostat or the co-administered active substance being necessary.
No dose adjustment is necessary for febuxostat when administered with hydrochlorothiazide.
No dose adjustment is necessary for warfarin when administered with febuxostat. Administration of
febuxostat (80 mg or 120 mg once daily) with warfarin had no effect on the pharmacokinetics of
warfarin in healthy subjects. INR and Factor VII activity were also not affected by the co-
administration of febuxostat.
Desipramine/CYP2D6 substrates.
Febuxostat was shown to be a weak inhibitor of CYP2D6
in vitro.
In a study in healthy subjects,
120 mg ADENURIC QD resulted in a mean 22% increase in AUC of desipramine, a CYP2D6
substrate indicating a potential weak inhibitory effect of febuxostat on the CYP2D6 enzyme
in vivo
.
Thus, co-administration of febuxostat with other CYP2D6 substrates is not expected to require any
dose adjustment for those compounds.
Antacids
Concomitant ingestion of an antacid containing magnesium hydroxide and aluminium hydroxide has
been shown to delay absorption of febuxostat (approximately 1 hour) and to cause a 32% decrease in
C
max
, but no significant change in AUC was observed. Therefore, febuxostat may be taken without
regard to antacid use.
17
Pregnancy
Data on a very limited number of exposed pregnancies have not indicated any adverse effects of
febuxostat on pregnancy or on the health of the foetus/new born child. Animal studies do not indicate
direct or indirect harmful effects with respect to pregnancy,
embryonal/foetal development or
parturition (see section 5.3).
The potential risk for human is unknown. Febuxostat should not be used
during pregnancy.
Breast feeding
It is unknown whether febuxostat is excreted in human breast milk. Animal studies have shown
excretion of this active substance in breast milk and an impaired development of suckling pups. A risk
to a suckling infant cannot be excluded. Febuxostat should not be used while breast-feeding.
No studies on the effects on the ability to drive and use machines have been performed. As with other
xanthine oxidase inhibitors adverse reactions such as somnolence, dizziness and paraesthesia have
been reported. Patients should exercise caution before driving, using machinery or participating in
dangerous activities until they are reasonably certain that ADENURIC does not adversely affect
performance.
A total of 4,072 subjects received at least one dose of ADENURIC (10 mg – 300 mg) in clinical
studies.
Combined phase 3 randomised controlled studies
In randomised controlled phase 3 clinical studies, >2,500 patients have been treated with 40 mg to
120 mg (1513 subject enrolled in a 26-week study (CONFIRMS), 536 subjects enrolled in a 28-week
study (APEX) and 507 subjects enrolled in a 52-weeks study (FACT)). The treatment-related events
(ADRs) were mostly mild or moderate in severity.
The most commonly reported ADRs (investigator assessment) are liver function abnormalities (5.0%),
diarrhoea (2.7%), nausea (1.3%), headache (1.2%), rash (1.2%).
Gout flares were also commonly observed soon after the start of treatment and during the first months.
Thereafter, the frequency of gout flare decreases in a time-dependent manner. As with other urate
lowering medicinal products, gout flare prophylaxis is recommended (see section 4.2 and 4.4).
A numerical greater incidence of investigator-reported cardiovascular APTC events (defined endpoints
from the Anti-Platelet Trialists’ Collaboration (APTC) including cardiovascular death, non-fatal
myocardial infarction, non-fatal stroke) was observed in the febuxostat total group compared to the
allopurinol group in the APEX and FACT studies (1.3 vs. 0.3 events per 100 PYs), but not in the
CONFIRMS study. The incidence of investigator-reported cardiovascular APTC events in the
combined Phase III studies (APEX, FACT and CONFIRMS studies) was 0.7 vs. 0.6 events per 100
PYs. In the long-term extension studies the incidences of investigator-reported APTC events were 1.2
and 0.6 events per 100 PYs for febuxostat and allopurinol, respectively. No statistically significant
differences were found and no causal relationship with febuxostat was established. Identified risk
factors among these patients were a medical history of atherosclerotic disease and/or myocardial
infarction, or of congestive heart failure.
Common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100) and rare (≥ 1/10,000 to < 1/1000)
adverse reactions suspected (investigator assessment) to be drug related occurring in patients treated
with 40 mg to 120 mg febuxostat and reported more than once in the total febuxostat treatment group
are listed below.
18
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Table 1: Treatment related adverse reactions in combined phase 3 randomized controlled studies
Blood and lymphatic system
disorders
Rare
Pancytopenia
Metabolism and nutrition
disorders
Common***
Gout flares
Uncommon
Decrease appetite
Rare
Weight increase/decrease, increase appetite, anorexia,
hyperlipidemia
Psychiatric disorders
Uncommon
Libido decreased, insomnia
Rare
Nervousness
Nervous system disorders
Common
Headache
Uncommon
Dizziness, paraesthesia, somnolence, altered taste, hypoaesthesia
Ear and labyrinth disorders
Rare
Tinnitus
Cardiac disorders
Uncommon
Atrial fibrillation, palpitations, ECG abnormal
Vascular disorders
Uncommon
Hypertension, flushing, hot flush
Respiratory system disorders
Uncommon
Dyspnoea, upper respiratory tract infection
Gastrointestinal disorders
Common
Diarrhoea
*
, nausea
Uncommon:
Abdominal pain, gastro-oesophageal reflux disease, vomiting, dry
mouth, dyspepsia, constipation, frequent
stools, flatulence,
gastrointestinal discomfort
Rare
Pancreatitis, mouth ulceration
Hepato-biliary disorders
Common
Liver function abnormalities*
Skin and subcutaneous tissue
disorders
Common
Rash
**
Uncommon
Dermatitis, urticaria, pruritus
Rare
Alopecia, hyperhidrosis
Musculoskeletal and connective
tissue disorders
Uncommon
Arthralgia, myalgia, musculoskeletal pain, muscle weakness,
muscle spasm
19














Rare
Arthritis, joint stiffness, musculoskeletal stiffness
Renal and urinary disorders
Uncommon
Nephrolithiasis, haematuria, pollakiuria, renal failure
Rare
Micturition urgency
Reproductive system and breast
disorder
Rare
Erectile dysfunction
General disorders and
administration site conditions
Uncommon
Fatigue, oedema, chest pain, chest discomfort
Rare
Thirst
Investigations
Uncommon
Blood amylase increase, platelet count decrease, blood creatinine
increase, haemoglobin decrease, blood urea increase, blood
triglycerides increase, blood cholesterol increase, haematocritic
decrease, blood lactate dehydrogenase increased
Rare
Blood glucose increased, activated partial thromboplastin time
prolonged, red blood cell count decrease, blood alkaline
phosphatase increase
*
Treatment-emergent non-infective diarrhoea and abnormal liver function tests in the combined
Phase III studies are more frequent in patients concomitantly treated with colchicine.
**
No serious rashes or severe hypersensitivity reactions were noted in the clinical studies.
*** See section 5.1 for incidences of gout flares in the individual Phase 3 randomized
controlled studies.
Long-term open label extension studies
In the long-term open label extension studies (1143 patients), the number of patients treated with
febuxostat 40 mg/80 mg/120 mg up to 1 year was 909, up to 2 years was 781, up to 3 years was 348,
and up to 4 and 5 years was 60. The treatment-related events reported during the long-term extension
studies were similar to those reported in the Phase 3 studies (see Table 1). The most commonly
reported treatment-related events (investigator assessment) are: liver function abnormalities, diarrhoea,
headache, rash, hypertension, oedema.
The following treatment-related events were reported more than once in the total febuxostat treatment
group and were reported as uncommon in patients taking febuxostat 40 mg/80 mg/120 mg in long-
term extension studies (up to 5 years, >2,660 Patient-years of exposure). These treatment-related
events were either not reported or reported at a lower frequency for these doses, in the combined Phase
3 studies: abdominal distension, cholelithiasis, bronchitis, weight increase, blood creatine increase,
diabetes mellitus, hyperlipidaemia, arthritis, muscle tightness, hyposmia, hemiparesis, cough, skin
discolouration, skin lesion, bursitis, proteinuria, petechiae, erectile dysfunction, blood potassium
increase, blood TSH increase, lymphocyte count decreased, WBC decrease.
No case of overdose has been reported. Patients with an overdose should be managed by symptomatic
and supportive care.
20








5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group:
Preparations inhibiting uric acid production,
ATC code: M04AA03
Mechanism of action
Uric acid is the end product of purine metabolism in humans and is generated in the cascade of
hypoxanthine → xanthine → uric acid. Both steps in the above transformations are catalyzed by
xanthine oxidase (XO). Febuxostat is a 2-arylthiazole derivative that achieves its therapeutic effect of
decreasing serum uric acid by selectively inhibiting XO. Febuxostat is a potent, non-purine selective
inhibitor of XO (NP-SIXO) with an
in vitro
inhibition Ki value less than one nanomolar. Febuxostat
has been shown to potently inhibit both the oxidized and reduced forms of XO. At therapeutic
concentrations febuxostat does not inhibit other enzymes involved in purine or pyrimidine
metabolism, namely, guanine deaminase, hypoxanthine guanine phosphoribosyltransferase, orotate
phosphoribosyltransferase, orotidine monophosphate decarboxylase or purine nucleoside
phosphorylase.
Clinical studies results
The efficacy of ADENURIC was demonstrated in three Phase 3 pivotal studies (the two pivotal APEX
and FACT studies, and the additional CONFIRMS study, described below) that were conducted in
4101 patients with hyperuricemia and gout. In each phase 3 pivotal study, ADENURIC demonstrated
superior ability to lower and maintain serum uric acid levels compared to allopurinol. The primary
efficacy endpoint in the APEX and FACT studies was the proportion of patients whose last 3 monthly
serum uric acid levels were < 6.0 mg/dl (357 µmol/l). In the additional phase 3 CONFIRMS study, for
which results became available after the marketing authorisation for ADENURIC was first issued, the
primary efficacy endpoint was the proportion of patients whose serum urate level was < 6.0 mg/dL at
the final visit. No patients with organ transplant have been included in these studies (see section 4.2).
APEX Study
: The Allopurinol and Placebo-Controlled Efficacy Study of Febuxostat (APEX) was a
Phase 3, randomized, double-blind, multicenter, 28-week study. One thousand and seventy-two (1072)
patients were randomized: placebo (n=134), ADENURIC 80 mg QD (n=267), ADENURIC 120 mg
QD (n=269), ADENURIC 240 mg QD (n=134) or allopurinol (300 mg QD [n=258] for patients with a
baseline serum creatinine ≤1.5 mg/dl or 100 mg QD [n=10] for patients with a baseline serum
creatinine >1.5 mg/dl and ≤2.0 mg/dl). Two hundred and forty mg febuxostat (2 times the
recommended highest dose) was used as a safety evaluation dose.
The APEX study showed statistically significant superiority of both the ADENURIC 80 mg QD and
the ADENURIC 120 mg QD treatment arms
versus
the conventionally used doses of allopurinol
300mg (n = 258) /100mg (n = 10) treatment arm in reducing the sUA below 6 mg/dl (357 µmol/l)
(see Table 2 and Figure 1).
FACT Study
: The Febuxostat Allopurinol Controlled Trial (FACT) Study was a Phase 3, randomized,
double-blind, multicenter, 52-week study. Seven hundred sixty (760) patients were randomized:
ADENURIC 80 mg QD (n=256), ADENURIC 120 mg QD (n=251), or allopurinol 300 mg QD
(n=253).
The FACT study showed the statistically significant superiority of both ADENURIC 80 mg and
ADENURIC 120 mg QD treatment arms
versus
the conventionally used dose of allopurinol 300 mg
treatment arm in reducing and maintaining sUA below 6 mg/dl (357 µmol/l).
21
Table 2 summarises the primary efficacy endpoint results:
Table 2
Proportion of Patients with Serum Uric Acid Levels <6.0 mg/dl (357µmol/l)
Last Three Monthly Visits
Study
ADENURIC
80 mg QD
ADENURIC
120 mg QD
Allopurinol
300 /
100 mg QD
1
APEX
(28 weeks)
48%
*
(n=262)
65%
*, #
(n=269)
22%
(n=268)
FACT
(52 weeks)
53%
*
(n=255)
62%
*
(n=250)
21%
(n=251)
22%
(n=519)
1
results from subjects receiving either 100 mg QD (n=10:
patients with
serum creatinine >1.5 and ≤2.0 mg/dl) or 300 mg QD (n=509) were pooled
for analyses.
* p < 0.001 vs allopurinol,
#
p < 0.001 vs 80 mg
51%
*
(n=517)
63%
*, #
(n=519)
The ability of ADENURIC to lower serum uric acid levels was prompt and persistent. Reduction in
serum uric acid level to <6.0 mg/dl (357 µmol/l) was noted by the Week 2 visit and was maintained
throughout treatment. The mean serum uric acid levels over time for each treatment group from the
two pivotal Phase 3 studies are shown in Figure 1.
Figure 1 Mean Serum Uric Acid Levels in Combined Pivotal Phase 3 Studies
11
10
Placebo
9
8
7
Allopurinol
6
ADE
NU
RI
C
80
mg
5
4
ADENURIC 120 mg
3
2
ADENURIC 240 mg
BL 2 4 6 8 12 16 20 24 28 32 36 40 44 48 52
Week
BL=baseline SEM=standard error of the mean
Note:
509 patients received allopurinol 300 mg QD; 10 patients with serum creatinine >1.5 and < 2.0
mg/dl were dosed with 100 mg QD. (10 patients out of 268 in APEX study).
240 mg febuxostat was used to evaluate the safety of febuxostat at twice the recommended
highest dose.
22
Combined
Results






































































































































































































































CONFIRMS Study: The CONFIRMS study was a Phase 3, randomized, controlled, 26-week study to
evaluate the safety and efficacy of febuxostat 40 mg and 80 mg, in comparison with allopurinol 300
mg or 200 mg, in patients with gout and hyperuricaemia. Two thousand and two hundred-sixty nine
(2269) patients were randomized: ADENURIC 40 mg QD (n=757), ADENURIC 80 mg QD (n=756),
or allopurinol 300/200 mg QD (n=756). At least 65% of the patients had mild-moderate renal
impairment (with creatinine clearance of 30-89 mL/min). Prophylaxis against gout flares was
obligatory over the 26-week period.
The proportion of patients with serum urate levels of < 6.0 mg/dL (357 µmol/L) at the final visit, was
45% for 40 mg febuxostat, 67% for febuxostat 80 mg and 42% for
allopurinol 300/200 mg,
respectively.
Primary endpoint in the sub-group of patients with renal impairment
The APEX Study evaluated efficacy in 40 patients with renal impairment (i.e., baseline serum
creatinine > 1.5 mg/dl and ≤2.0 mg/dl). For renally impaired subjects who were randomized to
allopurinol, the dose was capped at 100mg QD. ADENURIC achieved the primary efficacy endpoint
in 44% (80 mgQD), 45% (120 mg QD), and 60% (240 mg QD) of patients compared to 0% in the
allopurinol 100 mg QD and placebo groups.
There were no clinically significant differences in the percent decrease in serum uric acid
concentration in healthy subjects irrespective of their renal function (58 % in the normal renal function
group and 55% in the severe renal dysfunction group).
An analysis in patients with gout and renal impairment was prospectively defined in the CONFIRMS
study, and showed that febuxostat was significantly more efficacious in lowering serum urate levels to
< 6 mg/dL compared to allopurinol 300 mg/200 mg in patients who had gout with mild to moderate
renal impairment (65% of patients studied).
Primary endpoint in the sub group of patients with sUA ≥ 10 mg/dl
Approximately 40% of patients (combined APEX and FACT) had a baseline sUA of ≥ 10 mg/dl. In
this subgroup ADENURIC achieved the primary efficacy endpoint (sUA < 6.0 mg/dL at the last 3
visits) in 41% (80 mg QD), 48% (120 mg QD), and 66% (240 mg QD) of patients compared to 9% in
the allopurinol 300 mg/100 mg QD and 0 % in the placebo groups.
In the CONFIRMS study, the proportion of patients achieving the primary efficacy endpoint (sUA <
6.0 mg/dL at the final visit) for patients with a baseline serum urate level of ≥ 10 mg/dL treated with
febuxostat 40 mg QD was 27% (66/249), with febuxostat 80 mg QD 49% (125/254) and with
allopurinol 300 mg/200 mg QD 31% (72/230), respectively.
Clinical Outcomes: proportion of patients requiring treatment for a gout flare
Apex study: During the 8-week prophylaxis period, a greater proportion of subjects in the febuxostat
120 mg (36%) treatment group required treatment for gout flare compared to febuxostat 80 mg (28%),
allopurinol 300 mg (23%) and placebo (20%). Flares increased following the prophylaxis period and
gradually decreased over time. Between 46% and 55% of subjects received treatment for gout flares
from Week 8 and Week 28. Gout flares during the last 4 weeks of the study (Weeks 24-28) were
observed in 15% (febuxostat 80, 120 mg), 14% (allopurinol 300 mg) and 20% (placebo) of subjects.
Fact study: During the 8-week prophylaxis period, a greater proportion of subjects in the febuxostat
120 mg (36%) treatment group required treatment for a gout flare compared to both the febuxostat 80
mg (22%) and allopurinol 300 mg (21%) treatment groups. After the 8-week prophylaxis period, the
incidences of flares increased and gradually decreased over time (64% and 70% of subjects received
treatment for gout flares from Week 8-52). Gout flares during the last 4 weeks of the study (Weeks 49-
52) were observed in 6-8% (febuxostat 80 mg, 120 mg) and 11% (allopurinol 300 mg) of subjects.
The proportion of subjects requiring treatment for a gout flare (APEX and FACT Study) was
numerically lower in the groups that achieved an average post-baseline serum urate level <6.0 mg/dl,
<5.0 mg/dl, or <4.0 mg/dl compared to the group that achieved an average post-baseline serum urate
23
level ≥6.0 mg/dl during the last 32 weeks of the treatment period (Week 20-Week 24 to Week 49 - 52
intervals).
During the CONFIRMS study, the percentages of patients who required treatment for gout flares (Day
1 through Month 6) were 31% and 25% for the febuxostat 80 mg and allopurinol groups, respectively.
No difference in the proportion of patients requiring treatment for gout flares was observed between
the febuxostat 80 mg and 40 mg groups.
Long-term, open label extension Studies
EXCEL Study (C02-021): The Excel study was a three years Phase III, open label, multicenter,
randomised, allopurinol-controlled, safety extension study for patients who had completed the pivotal
Phase III studies (APEX or FACT). A total of 1086 patients were enrolled: ADENURIC 80 mg QD
(n=649), Adenuric 120 mg QD (n=292) and allopurinol 300/100 mg QD (n=145). About 69 % of
patients required no treatment change to achieve a final stable treatment. Patients who had 3
consecutive sUA levels >6.0 mg/dl were withdrawn.
Serum urate levels were maintained over time (i.e. 91% and 93% of patients on initial treatment with
febuxostat 80 mg and 120 mg, respectively, had sUA <6 mg/dl at Month 36).
Three years data showed a decrease in the incidence of gout flares with less than 4 % of patients
requiring treatment for a flare (i.e. more than 96 % of patients did not require treatment for a flare) at
Month 16-24 and at Month 30-36.
46% and 38%, of patients on final stable treatment of febuxostat 80 or 120 mg QD, respectively, had
complete resolution of the primary palpable tophus from baseline to the Final Visit.
FOCUS Study (TMX-01-005) was a 5 years Phase II, open-label, multicenter, safety extension study
for patients who had completed the febuxostat 4 weeks of double blind dosing in study TMX-00-004.
116 patients were enrolled and received initially febuxostat 80 mg QD. 62 % of patients required no
dose adjustment to maintain sUA <6 mg/dL and 38 % of patients required a dose adjustment to
achieve a final stable dose.
The proportion of patients with serum urate levels of <6.0 mg/dL (357 µmol/l) at the final visit was
greater than 80% (81-100%) at each febuxostat dose.
During the phase 3 clinical studies, mild liver function test abnormalities were observed in patients
treated with febuxostat (5.0%). These rates were similar to the rates reported on allopurinol (4.2%)
(see section 4.4). Increased TSH values (>5.5 µIU/ml) were observed in patients on long-term
treatment with febuxostat (5.5%) and patients with allopurinol (5.8%) in the long term open label
extension studies (see section 4.4).
5.2 Pharmacokinetic properties
In healthy subjects, maximum plasma concentrations (C
max
) and area under the plasma concentration
time curve (AUC) of febuxostat increased in a dose proportional manner following single and multiple
doses of 10 mg to 120 mg. For doses between 120 mg and 300 mg, a greater than dose proportional
increase in AUC is observed for febuxostat. There is no appreciable accumulation when doses of
10 mg to 240 mg are administered every 24 hours. Febuxostat has an apparent mean terminal
elimination half-life (t
1/2
) of approximately 5 to 8 hours.
Population pharmacokinetic/pharmacodynamic analyses were conducted in 211 patients with
hyperuricemia and gout, treated with ADENURIC 40-240 mg QD. In general, febuxostat
pharmacokinetic parameters estimated by these analyses are consistent with those obtained from
healthy subjects, indicating that healthy subjects are representative for
pharmacokinetic/pharmacodynamic assessment in the patient population with gout.
24
Absorption
Febuxostat is rapidly (t
max
of 1.0-1.5 h) and well absorbed (at least 84%). After single or multiple oral
80 and 120 mg once daily doses, C
max
is approximately 2.8-3.2 µg/ml, and 5.0-5.3 µg/ml, respectively.
Absolute bioavailability of the febuxostat tablet formulation has not been studied.
Following multiple oral 80 mg once daily doses or a single 120 mg dose with a high fat meal, there
was a 49% and 38% decrease in C
max
and a 18% and 16% decrease in AUC, respectively. However, no
clinically significant change in the percent decrease in serum uric acid concentration was observed
where tested (80 mg multiple dose). Thus, ADENURIC may be taken without regard to food.
Distribution
The apparent steady state volume of distribution (V
ss
/F) of febuxostat ranges from 29 to 75 l after oral
doses of 10-300 mg. The plasma protein binding of febuxostat is approximately 99.2%, (primarily to
albumin), and is constant over the concentration range achieved with 80 and 120 mg doses. Plasma
protein binding of the active metabolites ranges from about 82% to 91%.
Metabolism
Febuxostat is extensively metabolized by conjugation
via
uridine diphosphate glucuronosyltransferase
(UDPGT) enzyme system and oxidation
via
the cytochrome P450 (CYP) system. Four
pharmacologically active hydroxyl metabolites have been identified, of which three occur in plasma of
humans.
In vitro
studies with human liver microsomes showed that those oxidative metabolites were
formed primarily by CYP1A1, CYP1A2,
CYP2C8 or CYP2C9 and febuxostat glucuronide was formed mainly by UGT 1A1, 1A8, and 1A9
Elimination
Febuxostat is eliminated by both hepatic and renal pathways. Following an 80 mg oral dose of
14
C-
labeled febuxostat, approximately 49% of the dose was recovered in the urine as unchanged febuxostat
(3%), the acyl glucuronide of the active substance (30%), its known oxidative metabolites and their
conjugates (13%), and other unknown metabolites (3%). In addition to the urinary excretion,
approximately 45% of the dose was recovered in the faeces as the unchanged febuxostat (12%), the
acyl glucuronide of the active substance (1%), its known oxidative metabolites and their conjugates
(25%), and other unknown metabolites (7%).
Special populations
Renal insufficiency
Following multiple doses of 80 mg of ADENURIC in patients with mild, moderate or severe renal
insufficiency, the C
max
of febuxostat did not change, relative to subjects with normal renal function.
The mean total AUC of febuxostat increased by approximately 1.8-fold from 7.5 μg⋅h/ml in the
normal renal function group to 13.2 μg.h/ml in the severe renal dysfunction group. The C
max
and AUC
of active metabolites increased up to 2- and 4-fold, respectively. However, no dose adjustment is
necessary in patients with mild or moderate renal impairment.
Hepatic impairment
Following multiple doses of 80 mg of ADENURIC in patients with mild (Child-Pugh Class A) or
moderate (Child-Pugh Class B) hepatic impairment, the C
max
and AUC of febuxostat and its
metabolites did not change significantly compared to subjects with normal hepatic function. No
studies have been conducted in patients with severe hepatic impairment (Child-Pugh Class C).
Age
There were no significant changes observed in AUC of febuxostat or its metabolites following
multiple oral doses of ADENURIC in elderly as compared to younger healthy subjects.
Gender
Following multiple oral doses of ADENURIC, the C
max
and AUC were 24% and 12% higher in
females than in males, respectively. However, weight-corrected C
max
and AUC were similar between
the genders. No dose adjustment is needed based on gender.
25
5.3 Preclinical safety data
Effects in non-clinical studies were generally observed at exposures in excess of the maximum human
exposure.
Carcinogenesis, mutagenesis, impairment of fertility
In male rats, a statistically significant increase in urinary bladder tumours (transitional cell papilloma
and carcinoma) was found only in association with xanthine calculi in the high dose group, at
approximately 11 times human exposure. There was no significant increase in any other tumour type
in either male or female mice or rats. These findings are considered a consequence of species specific
purine metabolism and urine composition and of no relevance to clinical use.
A standard battery of test for genotoxicity did not reveal any biologically relevant genotoxic effects
for febuxostat.
Febuxostat at oral doses up to 48 mg/kg/day was found to have no effect on fertility and reproductive
performance of male and female rats.
There was no evidence of impaired fertility, teratogenic effects, or harm to the foetus due to
febuxostat. There was high dose maternal toxicity accompanied by a reduction in weaning index and
reduced development of offspring in rats at approximately 4.3 times human exposure. Teratology
studies, performed in pregnant rats at approximately 4.3 times and pregnant rabbits at approximately
13 times human exposure did not reveal any teratogenic effects.
6.
6.1 List of excipients
Tablet core
Lactose monohydrate
Microcrystalline cellulose
Magnesium stearate
Hydroxypropylcellulose
Croscarmellose sodium
Silica, colloidal hydrated
Tablet coating
Opadry II, Yellow, 85F42129 containing:
Polyvinyl alcohol
Titanium dioxide (E171)
Macrogols 3350
Talc
Iron oxide yellow (E172)
6.2 Incompatibilities
Not applicable.
6.3 Shelf life
3 years
6.4 Special precautions for storage
This medicinal product does not require any special storage condition.
26
6.5 Nature and contents of container
Clear (Aclar/PVC/Aluminium) blister of 14 tablets.
ADENURIC 120 mg is available in pack sizes of 14, 28, 42, 56, 84 and 98 film-coated tablets.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal
No special requirements.
7.
Menarini International Operations Luxembourg S.A.
1, Avenue de la Gare, L-1611 Luxembourg
Luxembourg
8.
EU/1/08/447/003
EU/1/08/447/004
EU/1/08/447/009
EU/1/08/447/010
EU/1/08/447/011
EU/1/08/447/012
9.
21/04/2008
Detailed information on this product is available on the website of the European Medicines Agency
27
A. AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
B. CONDITIONS OF THE MARKETING AUTHORISATION
28
ANNEX II
A. MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturer(s) responsible for batch release
Patheon France
40 Boulevard de Champaret
FR-38300 Bourgoin Jallieu
France
B. CONDITIONS OF THE MARKETING AUTHORISATION
•
Medicinal product subject to medical prescription.
•
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND
EFFECTIVE USE OF THE MEDICINAL PRODUCT
Not applicable
•
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, presented in Module 1.8.1. of the
Marketing Authorisation, is in place and functioning before and whilst the product is on the market.
Risk Management Plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in
the Pharmacovigilance Plan, as agreed in version 2.0 (19 February 2008) of the Risk Management
Plan (RMP) presented in Module 1.8.2. of the Marketing Authorisation Application and any
subsequent updates of the RMP agreed by the CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, the
updated RMP should be submitted at the same time as the next Periodic Safety Update Report
(PSUR).
In addition, an updated RMP should be submitted
•
When new information is received that may impact on the current Safety Specification,
Pharmacovigilance Plan or risk minimisation activities
•
Within 60 days of an important (pharmacovigilance or risk minimisation) milestone being
reached
•
At the request of the European Medicines Agency
29
ANNEX III
LABELLING AND PACKAGE LEAFLET
30
A. LABELLING
31
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
OUTER CARTON
1.
ADENURIC 80 mg film-coated tablets
Febuxostat
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each tablet contains 80 mg febuxostat.
3.
LIST OF EXCIPIENTS
Also contains lactose monohydrate.
See the package leaflet for further information.
4.
14 film-coated tablets
28 film-coated tablets
42 film-coated tablets
56 film-coated tablets
84 film-coated tablets
98 film-coated tablets
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
For oral use.
Read the package leaflet before use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
9.
SPECIAL STORAGE CONDITIONS
32



























10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Marketing Authorisation Holder:
Menarini International O. L. S.A.
1, Avenue de la Gare, L-1611 Luxembourg
Luxembourg
EU/1/08/447/001 28 film-coated tablets
EU/1/08/447/002 84 film-coated tablets
EU/1/08/447/005 14 film-coated tablets
EU/1/08/447/006 42 film-coated tablets
EU/1/08/447/007 56 film-coated tablets
EU/1/08/447/008 98 film-coated tablets
13. BATCH NUMBER
Lot:
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
ADENURIC 80 mg
33





















MINIMUM PARTICULARS TO APPEAR ON BLISTERS OR STRIPS
1.
ADENURIC 80 mg tablets
Febuxostat
2.
Menarini International O. L. S.A.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
Mon
Tue
Wed
Thurs
Fri
Sat
Sun
34
















PARTICULARS TO APPEAR ON THE OUTER PACKAGING
OUTER CARTON
1.
ADENURIC 120 mg film-coated tablets
Febuxostat
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each tablet contains 120 mg febuxostat.
3.
LIST OF EXCIPIENTS
Also contains lactose monohydrate.
See the package leaflet for further information.
4.
14 film-coated tablets
28 film-coated tablets
42 film-coated tablets
56 film-coated tablets
84 film-coated tablets
98 film-coated tablets
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
For oral use.
Read the package leaflet before use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
9.
SPECIAL STORAGE CONDITIONS
35




























10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Marketing Authorisation Holder:
Menarini International O. L. S.A.
1, Avenue de la Gare, L-1611 Luxembourg
Luxembourg
EU/1/08/447/003 28 film-coated tablets
EU/1/08/447/004 84 film-coated tablets
EU/1/08/447/009 14 film-coated tablets
EU/1/08/447/010 42 film-coated tablets
EU/1/08/447/011 56 film-coated tablets
EU/1/08/447/012 98 film-coated tablets
13. BATCH NUMBER
Lot:
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
ADENURIC 120 mg
36




















MINIMUM PARTICULARS TO APPEAR ON BLISTERS OR STRIPS
1.
ADENURIC 120 mg tablets
Febuxostat
2.
Menarini International O. L. S.A.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
Mon
Tue
Wed
Thurs
Fri
Sat
Sun
37
















B. PACKAGE LEAFLET
38
PACKAGE LEAFLET: INFORMATION FOR THE USER
ADENURIC 80 mg film-coated tablets
Febuxostat
Read all of this leaflet carefully before you start taking this medicine.
-
If you have any further questions, ask your doctor or pharmacist.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
-
If any of the side effects get serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor or pharmacist.
In this leaflet
:
1.
What ADENURIC is and what it is used for
2.
Before you take ADENURIC
3.
How to take ADENURIC
4.
How to store ADENURIC
6.
Further information
1.
WHAT ADENURIC IS AND WHAT IT IS USED FOR
ADENURIC tablets are used to treat gout, which is associated with an excess of a chemical called uric
acid (urate) in the body. In some people, the amount of uric acid builds up in the blood and may
become too high to remain soluble. When this happens, urate crystals may form in and around the
joints and kidneys. These crystals can cause sudden, severe pain, redness, warmth and swelling in a
joint (known as a gout attack). Left untreated, larger deposits called tophi (TOE-FI) may form in and
around joints. These tophi may cause joint and bone damage.
ADENURIC works by reducing uric acid levels. Keeping uric acid levels low by taking ADENURIC
once every day stops crystals building up, and over time it reduces symptoms. Keeping uric acid levels
sufficiently low for a long enough period can also shrink tophi.
2.
BEFORE YOU TAKE ADENURIC
Do not take ADENURIC if you are:
•
If you are allergic (hypersensitive) to febuxostat, the active ingredient of ADENURIC, or any of
the other ingredients in these tablets.
Take special care with ADENURIC
Tell your doctor before you start to take this medicine:
•
If you have or have had heart failure or heart problems
•
If you are being treated for high uric acid levels as a result of cancer disease or Lesch-Nyhan
syndrome (a rare inherited condition in which there is too much uric acid in the blood)
•
If you have thyroid problems
39
ADENURIC 120 mg film-coated tablets
Febuxostat
-
Keep this leaflet. You may need to read it again.
5
Possible side effects
If you are having a gout attack at the moment (a sudden onset of severe pain, tenderness, redness,
warmth and swelling in a joint), wait for the gout attack to subside before first starting treatment with
ADENURIC.
For some people, gout attacks may flare up when starting certain medicines that control uric acid
levels. Not everyone gets flares, but you could get a flare-up even if you are taking ADENURIC, and
especially during the first weeks or months of treatment. It is important to keep taking ADENURIC
even if you have a flare, as ADENURIC is still working to lower uric acid. Over time, gout flares will
occur less often and be less painful if you keep taking ADENURIC every day.
Your doctor will often prescribe other medicines, if they are needed, to help prevent or treat the
symptoms of flares (such as pain and swelling in a joint).
Your doctor may ask you to have blood tests to check that your liver is working normally.
Taking other medicines
Please tell your doctor or pharmacist if you are taking, or have recently taken, any other medicines,
including medicines obtained without a prescription.
It is especially important to tell your doctor or pharmacist if you are taking medicines containing any
of the following substances as they may interact with ADENURIC and your doctor may wish to
consider necessary measures:
•
Mercaptopurine (used to treat cancer)
•
Azathioprine (used to reduce immune response)
•
Theophylline (used to treat asthma)
Taking ADENURIC with food and drink
The tablets should be taken by mouth and can be taken with or without food.
Pregnancy and breast-feeding
It is not known if ADENURIC may harm your unborn child. Tell your doctor if you think you are
pregnant or if you are planning to become pregnant as ADENURIC should not be used during
pregnancy. It is not known if ADENURIC may pass into human breast milk. You should not use
ADENURIC if you are breast feeding, or if you are planning to breastfeed.
Driving and using machines
No studies on the effects of ADENURIC on the ability to drive and use machines have been
performed. However, you should be aware that you may experience dizziness, sleepiness and
numbness or tingling sensation during treatment and should not drive or operate machines if affected.
Important information about some of the ingredients of ADENURIC
ADENURIC tablets contain lactose (a type of sugar). If you have been told that you have an
intolerance to some sugars contact your doctor before taking this medicine.
3.
HOW TO TAKE ADENURIC
Always take ADENURIC exactly as your doctor has told you. You should check with your doctor or
pharmacist if you are not sure.
40
ADENURIC is available as either an 80 mg tablet or a 120 mg tablet. Your doctor will have prescribed
the strength most suitable for you.
•
The usual dose is one tablet daily. The back of the blister pack is marked with the days of the
week to help you check that you have taken a dose each day.
•
The tablets should be taken by mouth and can be taken with or without food.
It is important that you do not stop taking ADENURIC unless your doctor tells you to.
Continue to take ADENURIC every day even when you are not experiencing gout flare or attack.
If you take more ADENURIC than you should
In the event of an accidental overdose ask your doctor what to do, or contact your nearest accident and
emergency department.
If you forget to take ADENURIC
If you miss a dose of ADENURIC take it as soon as you remember unless it is almost time for your
next dose, in which case miss out the forgotten dose and take your next dose at the normal time. Do
not take a double dose to make up for a forgotten dose.
If you stop taking ADENURIC
Do not stop taking ADENURIC without the advice of your doctor even if you feel better. If you stop
taking ADENURIC your uric acid levels may begin to rise and your symptoms may worsen due to the
formation of new crystals of urate in and around your joints and kidneys.
If you have any further questions on the use of this product, ask your doctor or pharmacist.
4.
POSSIBLE SIDE EFFECTS
Like all medicines, ADENURIC can cause side effects, although not everybody gets them.
Common side effects
(reported in more than 1 in 100 patients but less than 1 in 10 patients) are:
•
abnormal liver test results
•
diarrhoea
•
headache
•
rashes
•
nausea
•
increase in gout symptoms
Uncommon side effects
(more than 1 in 1,000 patients but less than 1 in 100 patients) are:
•
decreased appetite, change in blood sugar levels (diabetes) of which a symptom may be
excessive thirst, increased blood fat levels
•
loss of sex drive
•
difficulty in sleeping, sleepiness
•
dizziness, numbness, tingling, reduced or altered sensation (hypoaesthesia or paraesthesia),
altered or reduced sense of taste
•
abnormal ECG heart tracing, irregular heartbeats, feeling your heart beat
•
hot flushes or blushing (e.g. redness of the face or neck), increased blood pressure
•
cough, shortness of breath, chest discomfort or pain, inflammation of nasal passage and/or throat
(upper respiratory tract infection), bronchitis
•
dry mouth, abdominal pain/discomfort or wind, heartburn/indigestion, constipation, more
frequent passing of stools, vomiting, stomach discomfort
41
•
itching, hives, skin inflammation or discolouration, other type of skin conditions
•
muscle cramp, muscle weakness, pain/ache in muscles/joints, bursitis or arthritis (inflammation
of joints usually accompanied by pain, swelling and/or stiffness), pain in extremity, back pain,
muscle spasm
•
blood in the urine, abnormal frequent urination, abnormal urine tests (increased level of proteins
in the urine), a reduction in the ability of the kidneys to function properly
•
fatigue, localised swelling due to the retention of fluids in the tissues (oedema), chest pain, chest
discomfort
•
Stones in the gallbladder or in bile ducts (cholelithiasis)
•
changes in blood chemistry or amount of blood cells or platelets (abnormal blood test results)
•
kidney stones
Rare side effects
(more than 1 in 10,000 patients but less than 1 in 1,000 patients) are:
•
nervousness
•
feeling thirsty
•
erectile difficulties
•
ringing in the ears
•
hair loss
•
mouth ulceration
•
inflammation of the pancreas: common symptoms are abdominal pain, nausea and vomiting
•
increased sweating
•
weight change (increase/decrease), increased appetite, uncontrolled loss of appetite (anorexia)
•
muscle and/or joint stiffness
•
abnormally low blood cell counts (white or red blood cells)
•
urgent need to urinate
If any of the side effects get serious, or if you notice any side effects not listed in this leaflet, please
tell your doctor or pharmacist.
5.
HOW TO STORE ADENURIC
•
Keep out of the reach and sight of children.
•
Do not use after the expiry date which is stated on the carton and the tablet blister foil after
‘EXP.’ The expiry date refers to the last day of that month.
•
This medicinal product does not require any special storage conditions.
Medicines should not be disposed of via wastewater or household waste. Ask your pharmacist how to
dispose of medicines no longer required. These measures will help to protect the environment.
6.
FURTHER INFORMATION
What ADENURIC contains
The active substance is febuxostat.
Each tablet contains 80 mg or 120 mg of febuxostat.
The other ingredients are:
Tablet core
: lactose monohydrate, microcrystalline cellulose, magnesium stearate,
hydroxypropylcellulose, croscarmellose sodium, colloidal hydrated silica.
Film-coating:
Opadry II yellow, 85F42129 containing: polyvinyl alcohol, titanium dioxide
(E171), macrogols 3350, talc, iron oxide yellow (E172)
42
What ADENURIC looks like and contents of the pack
ADENURIC film-coated tablets are pale yellow to yellow in colour and capsule shaped.
The 80 mg film-coated tablets are marked on one side with ‘80’. The 120 mg film-coated tablets are
marked on one side with ‘120’.
ADENURIC is supplied in 1 blisters of 14 tablets (14 tablet pack), 2 blisters of 14 tablets (28 tablet
pack), 3 blisters of 14 tablets (42 tablet pack), 4 blisters of 14 tablets (56 tablet pack), 6 blisters of 14
tablets (84 tablet pack), 6 blisters of 14 tablets (84 tablet pack) or 7 blisters of 14 tablets (98 tablet
pack). Not all pack sizes may be marketed.
Marketing Authorisation Holder and Manufacturer
Marketing Authorisation Holder
Menarini International Operations Luxembourg S.A.
1, Avenue de la Gare, L-1611 Luxembourg
Luxembourg
Patheon France
40 boulevard de Champaret
38300 Bourgoin Jallieu
France
For any information about this medicinal product, please contact the local representative of the
Marketing Authorisation Holder.
België/Belgique/Belgien
Menarini Benelux NV/SA
Tél/Tel: + 32 (0)2 721 4545
Luxembourg/Luxemburg
Menarini Benelux NV/SA
Tél/Tel: + 32 (0)2 721 4545
България
ТП “Берлин-Хеми АГ”
тел.: +359 2 96 55 365
Magyarország
Berlin-Chemie/A. Menarini Kft.
Tel.: +36 23501301
Česká republika
Berlin-Chemie/A.Menarini Ceska republika s.r.o.
Tel: +420 272 937 381
Malta
Menarini International Operations Luxembourg
S.A.
Tel: +352 264976
Danmark
Berlin-Chemie/A.Menarini Danmark ApS
Tlf: +4548 217 110
Nederland
Menarini Benelux NV/SA
Tel: +32 (0)2 721 4545
Deutschland
Berlin-Chemie AG
Tel: +49 (0) 30 67070
Norge
Menarini International Operations Luxembourg
S.A.
Tlf: +352 264976
Eesti
OÜ Berlin-Chemie Menarini Eesti
Tel: +372 667 5001
Österreich
A. Menarini Pharma GmbH.
Tel: +43 1 879 95 85-0
Ελλάδα
MENARINI HELLAS AE
Τηλ: +30 210 8316111-13
Polska
Berlin-Chemie/Menarini Polska Sp. z o.o.
Tel.: +48 22 566 21 00
43
anufacturer
España
Laboratorios Menarini S.A.
Tel: +34-93 462 88 00
Portugal
A. Menarini Portugal – Farmacêutica, S.A.
Tel: +351 210 935 500
France
MENARINI France
Tél: +33 (0)1 45 60 77 20
România
Berlin-Chemie Menarini Group
Tel: +40 211 232 34 32
Ireland
A. Menarini Pharmaceuticals Ltd
Tel: +353 1 284 6744
Slovenija
Berlin-Chemie AG, Podružnica Ljubljana
Tel: +386 01 300 2160
Ísland
Menarini International Operations Luxembourg
S.A.
Sími: +352 264976
Slovenská republika
Berlin-Chemie AG - obchodné zastúpenie v SR
Tel: +421 2 544 30 730
Italia
A. Menarini Industrie Farmaceutiche Riunite s.r.l.
Tel: +39-055 56801
Suomi/Finland
Berlin-Chemie/A.Menarini Suomi OY
Puh/Tel: +358 403 000 760
Κύπρος
MENARINI HELLAS AE
Τηλ: +30 210 8316111-13
Sverige
Menarini International Operations Luxembourg
S.A.
Tel: +352 264976
Latvija
SIA Berlin-Chemie/Menarini Baltic
Tel: +371 67103210
United Kingdom
A. Menarini Pharma U.K. S.R.L.
Tel: +44 (0)1628 856400
Lietuva
UAB “BERLIN-CHEMIE MENARINI BALTIC”
Tel: +370 52 691 947
This leaflet was last approved in
Detailed information on this medicine is available on the European Medicines Agency website
44
Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/adenuric.html
Copyright © 1995-2021 ITA all rights reserved.