










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
ADVATE 250 IU powder and solvent for solution for injection
2.
Each vial contains nominally 250 IU
*
human coagulation factor VIII (rDNA), octocog alfa
**
. After
reconstitution, each ml solution for injection contains approximately 50 IU octocog alfa.
*
The potency (International Units) is determined using the chromogenic assay against an in-house
standard that is referenced to the WHO standard. The specific activity is approximately 4,000-
10,000 IU/mg protein.
**
Human coagulation factor VIII produced by recombinant DNA technology in Chinese hamster
ovary (CHO) cells. Prepared without the addition of any (exogenous) human- or animal-derived
protein in the cell culture process, purification or final formulation.
Excipients: 0.45 mmol sodium (10 mg) per vial.
For a full list of excipients, see section 6.1.
3.
Powder and solvent for solution for injection
White to off-white friable powder.
After reconstitution, the solution is clear, colourless, free from foreign particles and has a pH of 6.7 to
7.3.
4.
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor VIII
deficiency).
ADVATE does not contain von Willebrand factor in pharmacologically effective quantities and is
therefore not indicated in von Willebrand disease.
Treatment should be initiated under the supervision of a physician experienced in the treatment
of haemophilia.
Posology
The posology and duration of the substitution therapy depend on the severity of the factor VIII
deficiency, on the location and extent of the bleeding and on the patient’s clinical condition.
On demand treatment
The dose of factor VIII (FVIII)is expressed in International Units (IU), which are related to the WHO
standard for factor VIII products. Factor VIII activity in plasma is expressed either as a percentage
(relative to normal human plasma) or in IUs (relative to the international standard for factor VIII in
plasma).
One IU of factor VIII activity is equivalent to that quantity of factor VIII in one ml of normal human
plasma. The calculation of the required dose of factor VIII is based on the empirical finding that 1 IU
2
factor VIII per kg body weight raises the plasma factor VIII activity by 2 IU/dl. The dose is
determined using the following formula:
Required units (IU) = body weight (kg) x desired factor VIII rise (%) x 0.5
In case of the following haemorrhagic events, the factor VIII activity should not fall below the given
plasma activity level (in % of normal or IU/dl) in the corresponding period. The followingtable 1 can
be used to guide dosing in bleeding episodes and surgery:
Table 1
Guide for dosing in bleeding episodes and surgery
Degree of haemorrhage/type of
surgical procedure
Factor VIII level
required (% or IU/dl)
Frequency of doses
(hours)/duration of therapy
(days)
Haemorrhage
Early haemarthrosis, muscle bleeding
or oral bleeding.
20 – 40
Repeat injections every 12 to 24
hours (8 to 24 hours for patients
under the age of 6) for at least 1
day, until the bleeding episode,
as indicated by pain, is resolved
or healing is achieved.
More extensive haemarthrosis, muscle
bleeding or haematoma.
30 – 60
Repeat injections every 12 to 24
hours (8 to 24 hours for patients
under the age of 6) for 3 – 4
days or more until pain and
acute disability are resolved.
Life-threatening haemorrhages.
60 – 100
Repeat injections every 8 to 24
hours (6 to 12 hours for patients
under the age of 6) until threat is
resolved.
Surgery
Minor
Including tooth extraction.
30 – 60
Every 24 hours (12 to 24 hours
for patients under the age of 6),
at least 1 day, until healing is
achieved.
Major
80 – 100
(pre- and postoperative)
Repeat injections every 8 to 24
hours (6 to 24 hours for patients
under the age of 6) until
adequate wound healing, then
continue therapy for at least
another 7 days to maintain a
factor VIII activity of 30% to
60% (IU/dl).
The dose and frequency of administration should be adapted to the clinical response in the individual
case. Under certain circumstances (e.g. presence of a low-titre inhibitor), doses larger than those
calculated using the formula may be necessary.
During the course of treatment, appropriate determination of plasma factor VIII levels is advised to
guide the dose to be administered and the frequency of repeated injections. In the case of major
surgical interventions in particular, precise monitoring of the substitution therapy by means of plasma
factor VIII activity assay is indispensable. Individual patients may vary in their response to factor
VIII, achieving different levels of
in vivo
recovery and demonstrating different half-lives.
3








Prophylaxis
For long-term prophylaxis against bleeding in patients with severe haemophilia A, the usual doses are
20 to 40 IU of factor VIIIper kg body weight at intervals of 2 to 3 days. In patients under the age of 6,
doses of 20 to 50 IU of factor VIII per kg body weight 3 to 4 times weekly are recommended.
Patients should be monitored for the development of factor VIII inhibitors. If the expected factor VIII
plasma activity levels are not attained, or if bleeding is not controlled with an appropriate dose, an
assay should be performed to determine if a factor VIII inhibitor is present. In patients with high levels
of inhibitor, factor VIII substitution therapy may not be effective and other therapeutic options should
be considered. The management of such patients should be directed by physicians with experience in
the care of patients with haemophilia (see section 4.4).
Method of administration
ADVATE should be administered via the intravenous route. In case of administration by a non health
care professional appropriate training is needed.
The rate of administration should be determined to ensure the comfort of the patient up to a maximum
of 10 ml/min.
In the interest of patients, it is recommended that every time ADVATE is administered, the name and
batch number of the product should be recorded.
For instructions for reconstitution prior to administration, see section 6.6.
Hypersensitivity to the active substance or to any of the excipients or to mouse or hamster proteins.
As with any intravenous protein product allergic type hypersensitivity reactions are possible. The
product contains traces of mouse and hamster proteins. Patients should be informed of the signs of
immediate-type hypersensitivity reactions including hives, pruritus, generalised urticaria, angioedema,
hypotension (e.g. dizziness or syncope), shock and acute respiratory distress (e.g. tightness in the
chest, wheezing). If these symptoms occur, they should be advised to discontinue use of the product
immediately and contact their physicians. In case of anaphylactic shock, the current medical standards
for shock treatment should be implemented (see section 4.8).
The formation of neutralising antibodies (inhibitors) against factor VIII is a known complication in the
management of individuals with haemophilia A. These inhibitors are usually IgGs directed against the
factor VIII procoagulant activity, which are quantified in Bethesda Units (BU) per ml of plasma using
the modified Bethesda assay. In patients who develop inhibitors to factor VIII, the condition may
manifest itself as an insufficient clinical response. In such cases, it is recommended that a specialised
haemophilia centre be contacted. The risk of developing inhibitors is correlated to the extent of
exposure to factor VIII, the risk being highest within the first 20 exposure days, and to other genetic
and environmental factors. Rarely, inhibitors may develop after the first 100 exposure days. Cases of
recurrent inhibitor (low titre) have been observed after switching from one factor VIII product to
another in previously treated patients with more than 100 exposure days who have a history of
inhibitor development. Patients treated with coagulation factor VIII should be carefully monitored for
the development of inhibitors by appropriate clinical observations and laboratory tests (see section
4.8).
After reconstitution this medicinal product contains 0.45 mmol sodium (10 mg) per vial. To be taken
into consideration by patients on a controlled sodium diet.
No interaction studies have been performed with ADVATE..
4
Animal reproduction studies have not been conducted with factor VIII. Based on the rare
occurrence
of haemophilia A in women, experience regarding the use of Factor VIII during pregnancy and
breast-feeding is not available. Therefore, factor VIII should be used during pregnancy and lactation
only if clearly indicated.
ADVATE has no influence on the ability to drive and use machines.
As with any intravenous protein product allergic type hypersensitivity reactions are possible. (see
section 4.4).
During clinical studies with ADVATE, a total of 56 adverse drug reactions (ADRs) were reported in
27 of 234 unique treated patients. The ADRs that occurred in the highest number of patients were
development of factor VIII inhibitors (5 patients), all occurring in previously untreated patients who
had elevated risk of inhibitor development, headache (5 patients), fever and dizziness (3 patients
each). Of the 56 ADRs, none were reported in neonates, 16 were reported in 13/32 infants, 7 were
reported in 4/56 children, 8 were reported in 4/31 adolescents and 25 were reported in 14/94 adults.
1
Frequency categories are defined according to the following convention: very common (≥1/10),
common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare
(<1/10,000), not known (cannot be calculated from the available data). Within each frequency
grouping, undesirable effects are presented in order of decreasing seriousness.
The following table 2 provides the frequency of adverse drug reactions in clinical trials:
Table 2
Frequency of adverse drug reactions (ADRs) in clinical trials
MedDRA Standard
System Organ Class
Adverse reaction
Patients
Adverse
Experience
Rate
(% patients)
a
Frequency
Infections and
infestations
Influenza
1
0.43
Uncommon
Laryngitis
1
0.43
Uncommon
Blood and lymphatic
system disorders
Lymphangitis
1
0.43
Uncommon
Nervous system
disorders
Headache
5
2.14
Common
Dizziness
3
1.28
Common
Memory impairment
1
0.43
Uncommon
Tremor
1
0.43
Uncommon
Migraine
1
0.43
Uncommon
Dysgeusia
1
0.43
Uncommon
Eye disorders
Eye inflammation
1
0.43
Uncommon
Vascular disorders
Haematoma
1
0.43
Uncommon
1
Neonates (age 0 – 1 month), infants (age 1 month – 2 years), children (age 2 - 12 years), adolescents (age 12 -
16 years) and adults (age over 16 years)
5
































Table 2
Frequency of adverse drug reactions (ADRs) in clinical trials
MedDRA Standard
Adverse reaction
Patients
Adverse
Frequency
System Organ Class
Experience
Rate
(% patients)
a
Hot flush
1
0.43
Uncommon
Pallor
1
0.43
Uncommon
Respiratory, thoracic and
mediastinal disorders
Dyspnoea
1
0.43
Uncommon
Gastrointestinal
disorders
Diarrhoea
2
0.85
Uncommon
Abdominal pain upper
1
0.43
Uncommon
Nausea
1
0.43
Uncommon
Vomiting
1
0.43
Uncommon
Skin and subcutaneous
tissue disorders
Pruritus
2
0.85
Uncommon
Rash
2
0.85
Uncommon
Hyperhidrosis
1
0.43
Uncommon
Dermatitis diaper
1
0.43
Uncommon
General disorders and
administration site
conditions
Pyrexia
3
1.28
Common
Oedema peripheral
1
0.43
Uncommon
Chest pain
1
0.43
Uncommon
Chills
1
0.43
Uncommon
Feeling abnormal
1
0.43
Uncommon
Investigations
Anti-factor VIII antibody
positive
5
2.14
Common
Alanine
aminotransferase
increased
1
0.43
Uncommon
Coagulation factor VIII
level decreased
b
1
0.43
Uncommon
Haematocrit decreased
1
0.43
Uncommon
Laboratory test abnormal
1
0.43
Uncommon
Injury, poisoning and
procedural complications
Post procedural
complication
1
0.43
Uncommon
Post procedural
haemorrhage
1
0.43
Uncommon
Procedural site reaction
1
0.43
Uncommon
a) Percent of patients calculated based on total number of unique patients (234).
b) The unexpected decrease in coagulation factor VIII levels occurred in one patient during
continuous infusion of ADVATE following surgery (postoperative days 10-14). Haemostasis
was maintained at all times during this period and both plasma factor VIII levels and clearance
rates returned to appropriate levels by postoperative day 15. Factor VIII inhibitor assays
performed after completion of continuous infusion and at study termination were negative.
6



























































Inhibitor Development
The immunogenicity of ADVATE was evaluated in previously treated patients. During clinical trials
haemophilia A (FVIII ≤ 2%), with previous exposure to factor VIII concentrates ≥ 150 days, one
patient developed a low-titre inhibitor (2.4 BU in the modified Bethesda assay) after 26 exposure days
to ADVATE. Follow-up inhibitor tests in this patient after withdrawal from the study were negative.
Also, in 53 paediatric patients under the age of 6, also diagnosed with severe to moderately severe
haemophilia A (FVIII ≤ 2%), with previous exposure to factor VIII concentrates ≥ 50 days, no FVIII
inhibitor was detected.
In previously untreated patients in an ongoing clinical study, 5 (20%) of 25 patients who received
ADVATE developed inhibitors to factor VIII. Of those, 4 were high-titre (≥ 5 BU) and 1 was low-titre
(< 5 BU). The frequency of FVIII inhibitors detected so far is within the expected and previously
observed range.
The patient’s immune response to trace amounts of contaminating proteins was analysed by examining
titres of antibodies to these proteins, laboratory parameters and reported adverse events. Of the 182
treated patients who were assessed for antibodies to Chinese hamster ovary (CHO) cell protein, 3
showed a statistically significant upward trend in titres by linear regression analysis and 4 displayed
sustained peaks or transient spikes. One patient had both a statistically significant upward trend and
displayed a sustained peak in anti-CHO protein antibody level, but no other signs or symptoms
indicative of an allergic or hypersensitivity response. Of the 182 treated patients who were assessed
for antibodies to murine IgG, 10 showed a statistically significant upward trend by linear regression
analysis and 2 displayed a sustained peak or transient spike. One patient had both a statistically
significant upward trend and displayed a sustained peak in anti-murine IgG antibody level. Four of
these patients reported isolated events of urticaria, pruritus, rash, and slightly elevated eosinophil
counts amongst numerous repeated exposures to the study product.
As with other intravenous products, allergic type hypersensitivity reactions, including
anaphylaxis/anaphylactoid reactions, have been reported with ADVATE (frequency not known).
No symptoms of overdose with recombinant coagulation factor VIII have been reported.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antihaemorrhagics, blood coagulation factor VIII. ATC code: B02BD02.
The factor VIII/von Willebrand Factor complex consists of two molecules (factor VIII and von
Willebrand factor) with different physiological functions.ADVATE contains recombinant coagulation
factor VIII (octocog alfa), a glycoprotein that is biologically equivalent to the factor VIII glycoprotein
found in human plasma.
Octocog alfa is a glycoprotein consisting of 2332 amino acids with an approximate molecular mass of
280 kD. When infused into a haemophilia patient, octocog alfa binds to endogenous von Willebrand
Factor in the patient’s circulation. Activated factor VIII acts as a Cofactor for activated Factor IX,
accelerating the conversion of Factor X to activated Factor X. Activated Factor X converts
prothrombin to thrombin. Thrombin then converts fibrinogen into fibrin and a clot can be formed.
Haemophilia A is a sex-linked hereditary disorder of blood coagulation due to decreased levels of
factor VIII activity and results in profuse bleeding into joints, muscles or internal organs, either
2
paediatric patients (age 0 –16 years) and adult patients (age over 16 years)
7
spontaneously or as a result of accidental or surgical trauma. The plasma levels of factor VIII are
increased by replacement therapy, thereby enabling a temporary correction of the factor VIII
deficiency and correction of the bleeding tendency.
5.2 Pharmacokinetic properties
All pharmacokinetic studies with ADVATE were conducted in previously treated patients with severe
to moderately severe haemophilia A (baseline factor VIII ≤ 2%). The analysis of plasma samples was
conducted in a central laboratory using a one-stage clotting assay. The pharmacokinetic parameters
derived from a crossover study of ADVATE in 100 previously treated patients ≥ 10 years of age are
listed in table 3 below.
Table 3
Summary of pharmacokinetic parameters for ADVATE in 100 patients with severe to
moderately severe haemophilia A (factor VIII ≤ 2%)
PK Parameter
Mean
SD
Median
Interquartile Range
AUC
0-∞
(IU·h/dl)
1527
*
528
1549
668
t
1/2
(h)
11.8
3.1
11.1
2.8
Adjusted Recovery
(IU/dl
/
IU/kg)
2.38
*
0.50
2.42
0.68
C
max
(IU/dl)
120
*
26
119
38
Clearance (dl/kg·h)
0.037
0.030
0.033
0.0147
MRT (h)
15.2
4.8
14.1
4.2
V
SS
(dl/kg)
0.52
0.39
0.48
0.13
* Geometric mean
Data from single-dose administrations of ADVATE in 53 paediatric patients less than 6 years of age
were used to obtain this table.
Table 4
Summary of pharmacokinetic parameters for
ADVATE in 53 paediatric patients with severe to moderately
severe haemophilia A
PK Parameter
Mean
SD
AUC
0-∞
(IU·h/dl)
1195
*
430
t
1/2
(h)
9.7
1.9
Adjusted Recovery
(IU/dl
/
IU/kg)
1.84
*
0.42
C
max
(IU/dl)
93
*
22
Clearance (dl/kg·h)
0.044
0.014
MRT (h)
12.2
3.1
V
SS
(dl/kg)
0.51
0.12
*
Geometric Mean
Adjusted recovery and terminal half-life (
t1/2
) was approximately 20% lower in young children (less
than 6 years of age) than in adults, which may be due in part to the known higher plasma volume per
kilogram body weight in younger patients.
Pharmacokinetic data with ADVATE on previously untreated patients are currently not available.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on studies of safety pharmacology, acute
toxicology, repeated dose toxicity, local toxicity and genotoxicity.
8





























6.
6.1 List of excipients
Powder
Mannitol
Sodium chloride
Histidine
Trehalose
Calcium chloride
Trometamol
Polysorbate 80
Glutathione (reduced)
Solvent
Sterilised water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products or solvents.
6.3 Shelf life
Unopened powder vial
2 years.
After reconstitution
Chemical and physical in-use stability has been demonstrated for 3 hours at 25°C. From a
microbiological point of view, the product should be used immediately after reconstitution.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C). Do not freeze.
During the shelf life, the product may be kept at room temperature (up to 25°C) for a single period not
exceeding 6 months. The beginning of storage at room temperature should be recorded on the product
carton. The product may not be returned to refrigerated storage again.
Keep the vial in the outer carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
6.5 Nature and contents of container
Each pack contains a powder vial, a vial containing 5 ml solvent (both type I glass closed with
chlorobutyl rubber stoppers) and a device for reconstitution (BAXJECT II)
.
6.6 Special precautions for disposal and other handling
Advate is to be administered intravenously after reconstitution of the lyophilized product with the
provided sterilised water for injections. After reconstitution the solution should be clear, colourless
and free from foreign particles.
-
For reconstitution use only the sterilised water for injections and the reconstitution device
provided in the pack. For administration the use of a luer-lock syringe is required.
9
-
Use within three hours after reconstitution.
-
Dispose any unused product or waste material in accordance with local requirements.
-
Do not use if the BAXJECT II device, its sterile barrier system or its packaging is damaged or
shows any sign of deterioration.
Reconstitution
Aseptic Technique should be used
1.
If the product is still stored in a refrigerator, take both the ADVATE powder and solvent vials
from the refrigerator and let them reach room temperature (between 15°C and 25°C).
2.
Wash your hands thoroughly using soap and warm water.
3.
Remove caps from powder and solvent vials.
4.
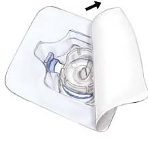
Open the package of BAXJECT II device by peeling away the paper lid without touching the
inside (Fig. a). Do not remove the device from the package. Do not use if the BAXJECT II
device, its sterile barrier system or its packaging is damaged or shows any sign of deterioration.
6.
Turn the package over and insert the clear plastic spike through the solvent stopper. Grip the
package at its edge and pull the package off BAXJECT II (Fig. b). Do not remove the blue cap
from the BAXJECT II device.
7.
For reconstitution only the water for injections and the reconstitution device provided in the
pack should be used. With BAXJECT II attached to the solvent vial, invert the system so that
the solvent vial is on top of the device. Insert the white plastic spike through the ADVATE
stopper. The vacuum will draw the solvent into the ADVATE vial (Fig. c).
8.
Swirl gently until all material is dissolved. Be sure that ADVATE is completely dissolved,
otherwise not all reconstituted solution will pass through the device filter. The product dissolves
rapidly (usually in less than 1 minute). After reconstitution the solution should be clear,
colourless and free from foreign particles.
Fig. a
Fig. b
Fig. c
Administration
Use Aseptic Technique
Parenteral medicinal products should be inspected for particulate matter prior to administration,
whenever solution and container permit. Only a clear and colourless solution should be used.
1.
Remove the blue cap from BAXJECT II. DO NOT DRAW AIR INTO THE SYRINGE.
Connect the syringe to BAXJECT II (Fig. d).
2.
Invert the system (the vial with the reconstituted solution has to be on top). Draw the
reconstituted solution into the syringe by pulling the plunger back slowly (Fig. e).
3.
Disconnect the syringe.
4.
Attach a butterfly needle to the syringe. Inject intravenously. The solution should be
administered slowly, at a rate as determined by the patient’s comfort level, not to exceed 10 ml
per minute. The pulse rate should be determined before and during administration of ADVATE.
Should a significant increase occur, reducing the rate of administration or temporarily
10
-
Do not refrigerate the preparation after reconstitution.
5.
Cleanse stoppers with alcohol swabs. Place the vials on a flat clean surface.




interrupting the injection usually allows the symptoms to disappear promptly (see sections 4.4
and 4.8).
Fig. d
Fig. e
7.
Baxter AG
Industriestrasse 67
A-1221 Vienna
Austria
8.
EU/1/03/271/001
9.
Date of first authorisation: 02 March 2004
Date of last renewal: 02 March 2009
Detailed information on this product is available on the website of the European Medicines Agency
(EMEA)
http://www.emea.europa.eu/
11


1.
ADVATE 500 IU powder and solvent for solution for injection.
2.
Each vial contains nominally 500 IU
*
human coagulation factor VIII (rDNA), octocog alfa
**
. After
reconstitution, each ml solution for injection contains approximately 100 IU octocog alfa.
*
The potency (International Units) is determined using the chromogenic assay against an in-house
standard that is referenced to the WHO standard. The specific activity is approximately 4,000-
10,000 IU/mg protein.
**
Human coagulation factor VIII produced by recombinant DNA technology in Chinese hamster
ovary (CHO) cells. Prepared without the addition of any (exogenous) human- or animal-derived
protein in the cell culture process, purification or final formulation.
Excipients: 0.45 mmol sodium (10 mg) per vial.
For a full list of excipients, see section 6.1.
3.
Powder and solvent for solution for injection.
White to off-white friable powder.
After reconstitution, the solution is clear, colourless, free from foreign particles and has a pH of 6.7 to
7.3.
4.
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor VIII
deficiency).
ADVATE does not contain von Willebrand Factor in pharmacologically effective quantities and is
therefore not indicated in von Willebrand disease.
Treatment should be initiated under the supervision of a physician experienced in the treatment
of haemophilia.
Posology
The posology and duration of the substitution therapy depend on the severity of the factor VIII
deficiency, on the location and extent of the bleeding and on the patient’s clinical condition.
On demand treatment
The dose of factor VIII (FVIII)is expressed in International Units (IU), which are related to the WHO
standard for factor VIII products. Factor VIII activity in plasma is expressed either as a percentage
(relative to normal human plasma) or in IUs (relative to the international standard for factor VIII in
plasma).
12
One IU of factor VIII activity is equivalent to that quantity of factor VIII in one ml of normal human
plasma. The calculation of the required dose of factor VIII is based on the empirical finding that 1 IU
factor VIII per kg body weight raises the plasma factor VIII activity by 2 IU/dl. The dose is
determined using the following formula:
Required units (IU) = body weight (kg) x desired factor VIII rise (%) x 0.5
In case of the following haemorrhagic events, the factor VIII activity should not fall below the given
plasma activity level (in % of normal or IU/dl) in the corresponding period. The followingtable 1 can
be used to guide dosing in bleeding episodes and surgery:
Table 1
Guide for dosing in bleeding episodes and surgery
Degree of haemorrhage/type of
surgical procedure
Factor VIII level
required (% or IU/dl)
Frequency of doses
(hours)/duration of therapy
(days)
Haemorrhage
Early haemarthrosis, muscle bleeding
or oral bleeding.
20 – 40
Repeat injections every 12 to 24
hours (8 to 24 hours for patients
under the age of 6) for at least 1
day, until the bleeding episode,
as indicated by pain, is resolved
or healing is achieved.
More extensive haemarthrosis, muscle
bleeding or haematoma.
30 – 60
Repeat injections every 12 to 24
hours (8 to 24 hours for patients
under the age of 6) for 3 – 4
days or more until pain and
acute disability are resolved.
Life threatening haemorrhages.
60 – 100
Repeat injections every 8 to 24
hours (6 to 12 hours for patients
under the age of 6) until threat is
resolved.
Surgery
Minor
Including tooth extraction.
30 – 60
Every 24 hours (12 to 24 hours
for patients under the age of 6),
at least 1 day, until healing is
achieved.
Major
80 – 100
(pre- and postoperative)
Repeat injections every 8 to 24
hours (6 to 24 hours for patients
under the age of 6) until
adequate wound healing, then
continue therapy for at least
another 7 days to maintain a
factor VIII activity of 30% to
60% (IU/dl).
The dose and frequency of administration should be adapted to the clinical response in the individual
case. Under certain circumstances (e.g. presence of a low titre inhibitor) doses larger than those
calculated using the formula may be necessary.
During the course of treatment, appropriate determination of plasma factor VIII levels is advised to
guide the dose to be administered and the frequency of repeated injections. In the case of major
surgical interventions in particular, precise monitoring of the substitution therapy by means of plasma
13








factor VIII activity assay is indispensable. Individual patients may vary in their response to factor
VIII, achieving different levels of
in vivo
recovery and demonstrating different half-lives.
Prophylaxis
For long-term prophylaxis against bleeding in patients with severe haemophilia A, the usual doses are
20 to 40 IU of factor VIIIper kg body weight at intervals of 2 to 3 days. In patients under the age of 6,
doses of 20 to 50 IU of factor VIII per kg body weight 3 to 4 times weekly are recommended.
Patients should be monitored for the development of factor VIII inhibitors. If the expected factor VIII
plasma activity levels are not attained, or if bleeding is not controlled with an appropriate dose, an
assay should be performed to determine if a factor VIII inhibitor is present. In patients with high levels
of inhibitor, factor VIII substitution therapy may not be effective and other therapeutic options should
be considered. The management of such patients should be directed by physicians with experience in
the care of patients with haemophilia (see section 4.4).
Method of administration
ADVATE should be administered via the intravenous route. In case of administration by a non health
care professional appropriate training is needed.
The rate of administration should be determined to ensure the comfort of the patient up to a maximum
of 10 ml/min.
In the interest of patients, it is recommended that every time ADVATE is administered, the name and
batch number of the product should be recorded.
For instructions for reconstitution prior to administration, see section 6.6.
Hypersensitivity to the active substance or to any of the excipients or to mouse or hamster proteins.
As with any intravenous protein product allergic type hypersensitivity reactions are possible. The
product contains traces of mouse and hamster proteins. Patients should be informed of the signs of
immediate-type hypersensitivity reactions including hives, pruritus, generalised urticaria, angioedema,
hypotension (e.g. dizziness or syncope), shock and acute respiratory distress (e.g. tightness in the
chest, wheezing). If these symptoms occur, they should be advised to discontinue use of the product
immediately and contact their physicians. In case of anaphylactic shock, the current medical standards
for shock treatment should be implemented (see section 4.8).
The formation of neutralising antibodies (inhibitors) against factor VIII is a known complication in the
management of individuals with haemophilia A. These inhibitors are usually IgGs directed against the
factor VIII procoagulant activity, which are quantified in Bethesda Units (BU) per ml of plasma using
the modified Bethesda assay. In patients who develop inhibitors to factor VIII, the condition may
manifest itself as an insufficient clinical response. In such cases, it is recommended that a specialised
haemophilia centre be contacted. The risk of developing inhibitors is correlated to the extent of
exposure to factor VIII, the risk being highest within the first 20 exposure days, and to other genetic
and environmental factors. Rarely, inhibitors may develop after the first 100 exposure days. Cases of
recurrent inhibitor (low titre) have been observed after switching from one factor VIII product to
another in previously treated patients with more than 100 exposure days who have a history of
inhibitor development. Patients treated with coagulation factor VIII should be carefully monitored for
the development of inhibitors by appropriate clinical observations and laboratory tests (see section
4.8).
After reconstitution this medicinal product contains 0.45 mmol sodium (10 mg) per vial. To be taken
into consideration by patients on a controlled sodium diet.
14
No interaction studies have been performed with ADVATE.
Animal reproduction studies have not been conducted with factor VIII. Based on the rare occurrence
of haemophilia A in women, experience regarding the use of factor VIII during pregnancy and breast-
feeding is not available. Therefore, factor VIII should be used during pregnancy and lactation only if
clearly indicated.
ADVATE has no influence on the ability to drive and use machines.
As with any intravenous protein product allergic type hypersensitivity reactions are possible. (see
section 4.4).
During clinical studies with ADVATE, a total of 56 adverse drug reactions (ADRs) were reported in
27 of 234 unique treated patients. The ADRs that occurred in the highest number of patients were
development of factor VIII inhibitors (5 patients), all occurring in previously untreated patients who
had elevated risk of inhibitor development, headache (5 patients), fever and dizziness (3 patients
each). Of the 56 ADRs, none were reported in neonates, 16 were reported in 13/32 infants, 7 were
reported in 4/56 children, 8 were reported in 4/31 adolescents and 25 were reported in 14/94 adults.
3
Frequency categories are defined according to the following convention: very common (≥1/10),
common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare
(<1/10,000), not known (cannot be calculated from the available data). Within each frequency
grouping, undesirable effects are presented in order of decreasing seriousness.
The following table 2 provides the frequency of adverse drug reactions in clinical trials:
Table 2
Frequency of adverse drug reactions (ADRs) in clinical trials
MedDRA Standard
System Organ Class
Adverse reaction
Patients
Adverse
Experience
Rate
(% patients)
a
Frequency
Infections and
infestations
Influenza
1
0.43
Uncommon
Laryngitis
1
0.43
Uncommon
Blood and lymphatic
system disorders
Lymphangitis
1
0.43
Uncommon
Nervous system
disorders
Headache
5
2.14
Common
Dizziness
3
1.28
Common
Memory impairment
1
0.43
Uncommon
Tremor
1
0.43
Uncommon
Migraine
1
0.43
Uncommon
Dysgeusia
1
0.43
Uncommon
3
Neonates (age 0 – 1 month), infants (age 1 month – 2 years), children (age 2 - 12 years), adolescents (age 12 -
16 years) and adults (age over 16 years)
15




























Table 2
Frequency of adverse drug reactions (ADRs) in clinical trials
MedDRA Standard
Adverse reaction
Patients
Adverse
Frequency
System Organ Class
Experience
Rate
(% patients)
a
Eye disorders
Eye inflammation
1
0.43
Uncommon
Vascular disorders
Haematoma
1
0.43
Uncommon
Hot flush
1
0.43
Uncommon
Pallor
1
0.43
Uncommon
Respiratory, thoracic and
mediastinal disorders
Dyspnoea
1
0.43
Uncommon
Gastrointestinal
disorders
Diarrhoea
2
0.85
Uncommon
Abdominal pain upper
1
0.43
Uncommon
Nausea
1
0.43
Uncommon
Vomiting
1
0.43
Uncommon
Skin and subcutaneous
tissue disorders
Pruritus
2
0.85
Uncommon
Rash
2
0.85
Uncommon
Hyperhidrosis
1
0.43
Uncommon
Dermatitis diaper
1
0.43
Uncommon
General disorders and
administration site
conditions
Pyrexia
3
1.28
Common
Oedema peripheral
1
0.43
Uncommon
Chest pain
1
0.43
Uncommon
Chills
1
0.43
Uncommon
Feeling abnormal
1
0.43
Uncommon
Investigations
Anti-factor VIII antibody
positive
5
2.14
Common
Alanine
aminotransferase
increased
1
0.43
Uncommon
Coagulation factor VIII
level decreased
b
1
0.43
Uncommon
Haematocrit decreased
1
0.43
Uncommon
Laboratory test abnormal
1
0.43
Uncommon
Injury, poisoning and
procedural complications
Post procedural
complication
1
0.43
Uncommon
Post procedural
haemorrhage
1
0.43
Uncommon
Procedural site reaction
1
0.43
Uncommon
a) Percent of patients calculated based on total number of unique patients (234).
b) The unexpected decrease in coagulation factor VIII levels occurred in one patient during
continuous infusion of ADVATE following surgery (postoperative days 10-14). Haemostasis
was maintained at all times during this period and both plasma factor VIII levels and clearance
rates returned to appropriate levels by postoperative day 15. Factor VIII inhibitor assays
performed after completion of continuous infusion and at study termination were negative.
16































































Inhibitor Development
The immunogenicity of ADVATE was evaluated in previously treated patients. During clinical trials
haemophilia A (FVIII ≤ 2%), with previous exposure to factor VIII concentrates ≥ 150 days, one
patient developed a low-titre inhibitor (2.4 BU in the modified Bethesda assay) after 26 exposure days
to ADVATE. Follow-up inhibitor tests in this patient after withdrawal from the study were negative.
Also, in 53 paediatric patients under the age of 6, also diagnosed with severe to moderately severe
haemophilia A (FVIII ≤ 2%), with previous exposure to factor VIII concentrates ≥ 50 days, no FVIII
inhibitor was detected.
In previously untreated patients in an ongoing clinical study, 5 (20%) of 25 patients who received
ADVATE developed inhibitors to factor VIII. Of those, 4 were high-titre (≥ 5 BU) and 1 was low-titre
(< 5 BU). The frequency of FVIII inhibitors detected so far is within the expected and previously
observed range.
The patient’s immune response to trace amounts of contaminating proteins was analysed by examining
titres of antibodies to these proteins, laboratory parameters and reported adverse events. Of the 182
treated patients who were assessed for antibodies to Chinese hamster ovary (CHO) cell protein, 3
showed a statistically significant upward trend in titres by linear regression analysis and 4 displayed
sustained peaks or transient spikes. One patient had both a statistically significant upward trend and
displayed a sustained peak in anti-CHO protein antibody level, but no other signs or symptoms
indicative of an allergic or hypersensitivity response. Of the 182 treated patients who were assessed
for antibodies to murine IgG, 10 showed a statistically significant upward trend by linear regression
analysis and 2 displayed a sustained peak or transient spike. One patient had both a statistically
significant upward trend and displayed a sustained peak in anti-murine IgG antibody level. Four of
these patients reported isolated events of urticaria, pruritus, rash, and slightly elevated eosinophil
counts amongst numerous repeated exposures to the study product.
As with other intravenous products, allergic type hypersensitivity reactions, including
anaphylaxis/anaphylactoid reactions, have been reported with ADVATE (frequency not known).
No symptoms of overdose with recombinant coagulation factor VIII have been reported.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antihaemorrhagics, blood coagulation factor VIII. ATC code: B02BD02.
The factor VIII/von Willebrand Factor complex consists of two molecules (factor VIII and von
Willebrand factor) with different physiological functions. ADVATE contains recombinant coagulation
factor VIII (octocog alfa), a glycoprotein that is biologically equivalent to the factor VIII glycoprotein
found in human plasma.
Octocog alfa is a glycoprotein consisting of 2332 amino acids with an approximate molecular mass of
280 kD. When infused into a haemophilia patient, octocog alfa binds to endogenous von Willebrand
Factor in the patient’s circulation. Activated factor VIII acts as a Cofactor for activated Factor IX,
accelerating the conversion of Factor X to activated Factor X. Activated Factor X converts
prothrombin to thrombin. Thrombin then converts fibrinogen into fibrin and a clot can be formed.
Haemophilia A is a sex-linked hereditary disorder of blood coagulation due to decreased levels of
factor VIII activity and results in profuse bleeding into joints, muscles or internal organs, either
4
paediatric patients (age 0 –16 years) and adult patients (age over 16 years)
17
spontaneously or as a result of accidental or surgical trauma. The plasma levels of factor VIII are
increased by replacement therapy, thereby enabling a temporary correction of the factor VIII
deficiency and correction of the bleeding tendency.
5.2 Pharmacokinetic properties
All pharmacokinetic studies with ADVATE were conducted in previously treated patients with severe
to moderately severe haemophilia A (baseline factor VIII ≤ 2%). The analysis of plasma samples was
conducted in a central laboratory using a one-stage clotting assay. The pharmacokinetic parameters
derived from a crossover study of ADVATE in 100 previously treated patients ≥ 10 years of age are
listed in table 3 below.
Table 3
Summary of pharmacokinetic parameters for ADVATE in 100 patients with severe to
moderately severe haemophilia A (factor VIII ≤ 2%)
PK Parameter
Mean
SD
Median
Interquartile
Range
AUC
0-∞
(IU·h/dl)
1527
*
528
1549
668
t
1/2
(h)
11.8
3.1
11.1
2.8
Adjusted Recovery
(IU/dl
/
IU/kg)
2.38
*
0.50
2.42
0.68
C
max
(IU/dl)
120
*
26
119
38
Clearance (dl/kg·h)
0.037
0.030
0.033
0.0147
MRT (h)
15.2
4.8
14.1
4.2
V
SS
(dl/kg)
0.52
0.39
0.48
0.13
* Geometric mean
Data from single-dose administrations of ADVATE in 53 paediatric patients less than 6 years of age
were used to obtain this table.
Table 4
Summary of pharmacokinetic parameters for
ADVATE in 53 paediatric patients with severe to moderately
severe haemophilia A
PK Parameter
Mean
SD
AUC
0-∞
(IU·h/dl)
1195
*
430
t
1/2
(h)
9.7
1.9
Adjusted Recovery
(IU/dl
/
IU/kg)
1.84
*
0.42
C
max
(IU/dl)
93
*
22
Clearance (dl/kg·h)
0.044
0.014
MRT (h)
12.2
3.1
V
SS
(dl/kg)
0.51
0.12
*
Geometric Mean
Adjusted recovery and terminal half-life (
t1/2
) was approximately 20% lower in young children (less
than 6 years of age) than in adults, which may be due in part to the known higher plasma volume per
kilogram body weight in younger patients.
Pharmacokinetic data with ADVATE on previously untreated patients are currently not available.
18





























5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on studies of safety pharmacology, acute
toxicology, repeated dose toxicity, local toxicity and genotoxicity.
6.
6.1 List of excipients
Powder
Mannitol
Sodium chloride
Histidine
Trehalose
Calcium chloride
Trometamol
Polysorbate 80
Glutathione (reduced)
Solvent
Sterilised water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products or solvents.
6.3 Shelf life
Unopened powder vial
2 years.
After reconstitution
Chemical and physical in-use stability has been demonstrated for 3 hours at 25°C. From a
microbiological point of view, the product should be used immediately after reconstitution.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C). Do not freeze.
During the shelf life, the product may be kept at room temperature (up to 25°C) for a single period not
exceeding 6 months. The beginning of storage at room temperature should be recorded on the product
carton. The product may not be returned to refrigerated storage again.
Keep the vial in the outer carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
6.5 Nature and contents of container
Each pack contains a powder vial, a vial containing 5 ml solvent (both type I glass closed with
chlorobutyl rubber stoppers) and a device for reconstitution (BAXJECT II)
.
19
6.6 Special precautions for disposal and other handling
ADVATE is to be administered intravenously after reconstitution of the lyophilized product with the
provided sterilised water for injections. After reconstitution the solution should be clear, colourless
and free from foreign particles.
-
For reconstitution use only the sterilised water for injections and the reconstitution device
provided in the pack. For administration the use of a luer-lock syringe is required.
-
Use within three hours after reconstitution.
-
Dispose any unused product or waste material in accordance with local requirements.
-
Do not use if the BAXJECT II device, its sterile barrier system or its packaging is damaged or
shows any sign of deterioration.
Reconstitution
Aseptic Technique should be used
1.
If the product is still stored in a refrigerator, take both the ADVATE powder and solvent vials
from the refrigerator and let them reach room temperature (between 15°C and 25°C).
2.
Wash your hands thoroughly using soap and warm water.
3.
Cleanse stoppers with alcohol swabs. Place the vials on a flat clean surface.
5.
Open the package of BAXJECT II device by peeling away the paper lid without touching the
inside (Fig. a). Do not remove the device from the package. Do not use if the BAXJECT II
device, its sterile barrier system or its packaging is damaged or shows any sign of deterioration.
6.
Turn the package over and insert the clear plastic spike through the solvent stopper. Grip the
package at its edge and pull the package off BAXJECT II (Fig. b). Do not remove the blue cap
from the BAXJECT II device.
7.
For reconstitution only the water for injections and the reconstitution device provided in the
pack should be used. With BAXJECT II attached to the solvent vial, invert the system so that
the solvent vial is on top of the device. Insert the white plastic spike through the ADVATE
stopper. The vacuum will draw the solvent into the ADVATE vial (Fig. c).
8.
Swirl gently until all material is dissolved. Be sure that ADVATE is completely dissolved,
otherwise not all reconstituted solution will pass through the device filter. The product dissolves
rapidly (usually in less than 1 minute). After reconstitution the solution should be clear,
colourless and free from foreign particles.
Fig. a
Fig. b
Fig. c
Administration
Use Aseptic Technique
Parenteral medicinal products should be inspected for particulate matter prior to administration,
whenever solution and container permit. Only a clear and colourless solution should be used.
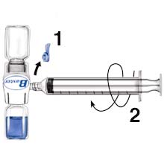
1.
Remove the blue cap from BAXJECT II. DO NOT DRAW AIR INTO THE SYRINGE.
Connect the syringe to BAXJECT II (Fig. d).
20
-
Do not refrigerate the preparation after reconstitution.
4.
Remove caps from powder and solvent vials.



2.
Invert the system (the vial with the reconstituted solution has to be on top). Draw the
reconstituted solution into the syringe by pulling the plunger back slowly (Fig. e).
3.
Disconnect the syringe.
4.
Attach a butterfly needle to the syringe. Inject intravenously. The solution should be
administered slowly, at a rate as determined by the patient’s comfort level, not to exceed 10 ml
per minute. The pulse rate should be determined before and during administration of ADVATE.
Should a significant increase occur, reducing the rate of administration or temporarily
interrupting the injection usually allows the symptoms to disappear promptly (see sections 4.4
and 4.8).
Fig. d
Fig. e
7.
Baxter AG
Industriestrasse 67
A-1221 Vienna
Austria
8.
EU/1/03/271/002
9.
Date of first authorisation: 02 March 2004
Date of last renewal: 02 March 2009
Detailed information on this product is available on the website of the European Medicines Agency
(EMEA)
http://www.emea.europa.eu/
21

1.
ADVATE 1000 IU powder and solvent for solution for injection.
2.
Each vial contains nominally 1000 IU
*
human coagulation factor VIII (rDNA), octocog alfa
**
. After
reconstitution, each ml solution for injection contains approximately 200 IU octocog alfa.
*
The potency (International Units) is determined using the chromogenic assay against an in-house
standard that is referenced to the WHO standard. The specific activity is approximately 4,000-
10,000 IU/mg protein.
**
Human coagulation factor VIII produced by recombinant DNA technology in Chinese hamster
ovary (CHO) cells. Prepared without the addition of any (exogenous) human- or animal-derived
protein in the cell culture process, purification or final formulation.
Excipients: 0.45 mmol sodium (10 mg) per vial.
For a full list of excipients, see section 6.1.
3.
Powder and solvent for solution for injection.
White to off-white friable powder.
After reconstitution, the solution is clear, colourless, free from foreign particles and has a pH of 6.7 to
7.3.
4.
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor VIII
deficiency).
ADVATE does not contain von Willebrand Factor in pharmacologically effective quantities and is
therefore not indicated in von Willebrand disease.
Treatment should be initiated under the supervision of a physician experienced in the treatment
of haemophilia.
Posology
The posology and duration of the substitution therapy depend on the severity of the factor VIII
deficiency, on the location and extent of the bleeding and on the patient’s clinical condition.
On demand treatment
The dose of factor VIII (FVIII)is expressed in International Units (IU), which are related to the WHO
standard for factor VIII products. Factor VIII activity in plasma is expressed either as a percentage
(relative to normal human plasma) or in IUs (relative to the international standard for factor VIII in
plasma).
22
One IU of factor VIII activity is equivalent to that quantity of factor VIII in one ml of normal human
plasma. The calculation of the required dose of factor VIII is based on the empirical finding that 1 IU
factor VIII per kg body weight raises the plasma factor VIII activity by 2 IU/dl. The dose is
determined using the following formula:
Required units (IU) = body weight (kg) x desired factor VIII rise (%) x 0.5
In case of the following haemorrhagic events, the factor VIII activity should not fall below the given
plasma activity level (in % of normal or IU/dl) in the corresponding period. The followingtable 1 can
be used to guide dosing in bleeding episodes and surgery:
Table 1
Guide for dosing in bleeding episodes and surgery
Degree of haemorrhage/type of
surgical procedure
Factor VIII level
required (% or IU/dl)
Frequency of doses
(hours)/duration of therapy
(days)
Haemorrhage
Early haemarthrosis, muscle bleeding
or oral bleeding.
20 – 40
Repeat injections every 12 to 24
hours (8 to 24 hours for patients
under the age of 6) for at least 1
day, until the bleeding episode,
as indicated by pain, is resolved
or healing is achieved.
More extensive haemarthrosis, muscle
bleeding or haematoma.
30 – 60
Repeat injections every 12 to 24
hours (8 to 24 hours for patients
under the age of 6) for 3 – 4
days or more until pain and
acute disability are resolved.
Life threatening haemorrhages.
60 – 100
Repeat injections every 8 to 24
hours (6 to 12 hours for patients
under the age of 6) until threat is
resolved.
Surgery
Minor
Including tooth extraction.
30 – 60
Every 24 hours (12 to 24 hours
for patients under the age of 6),
at least 1 day, until healing is
achieved.
Major
80 – 100
(pre- and postoperative)
Repeat injections every 8 to 24
hours (6 to 24 hours for patients
under the age of 6) until
adequate wound healing, then
continue therapy for at least
another 7 days to maintain a
factor VIII activity of 30% to
60% (IU/dl).
The dose and frequency of administration should be adapted to the clinical response in the individual
case. Under certain circumstances (e.g. presence of a low titre inhibitor) doses larger than those
calculated using the formula may be necessary.
During the course of treatment, appropriate determination of plasma factor VIII levels is advised to
guide the dose to be administered and the frequency of repeated injections. In the case of major
surgical interventions in particular, precise monitoring of the substitution therapy by means of plasma
23








factor VIII activity assay is indispensable. Individual patients may vary in their response to factor
VIII, achieving different levels of
in vivo
recovery and demonstrating different half-lives.
Prophylaxis
For long-term prophylaxis against bleeding in patients with severe haemophilia A, the usual doses are
20 to 40 IU of factor VIIIper kg body weight at intervals of 2 to 3 days. In patients under the age of 6,
doses of 20 to 50 IU of factor VIII per kg body weight 3 to 4 times weekly are recommended.
Patients should be monitored for the development of factor VIII inhibitors. If the expected factor VIII
plasma activity levels are not attained, or if bleeding is not controlled with an appropriate dose, an
assay should be performed to determine if a factor VIII inhibitor is present. In patients with high levels
of inhibitor, factor VIII substitution therapy may not be effective and other therapeutic options should
be considered. The management of such patients should be directed by physicians with experience in
the care of patients with haemophilia (see section 4.4).
Method of administration
ADVATE should be administered via the intravenous route. In case of administration by a non health
care professional appropriate training is needed.
The rate of administration should be determined to ensure the comfort of the patient up to a maximum
of 10 ml/min.
In the interest of patients, it is recommended that every time ADVATE is administered, the name and
batch number of the product should be recorded.
For instructions for reconstitution prior to administration, see section 6.6.
Hypersensitivity to the active substance or to any of the excipients or to mouse or hamster proteins.
As with any intravenous protein product allergic type hypersensitivity reactions are possible. The
product contains traces of mouse and hamster proteins. Patients should be informed of the signs of
immediate-type hypersensitivity reactions including hives, pruritus, generalised urticaria, angioedema,
hypotension (e.g. dizziness or syncope), shock and acute respiratory distress (e.g. tightness in the
chest, wheezing). If these symptoms occur, they should be advised to discontinue use of the product
immediately and contact their physicians. In case of anaphylactic shock, the current medical standards
for shock treatment should be implemented (see section 4.8).
The formation of neutralising antibodies (inhibitors) against factor VIII is a known complication in the
management of individuals with haemophilia A. These inhibitors are usually IgGs directed against the
factor VIII procoagulant activity, which are quantified in Bethesda Units (BU) per ml of plasma using
the modified Bethesda assay. In patients who develop inhibitors to factor VIII, the condition may
manifest itself as an insufficient clinical response. In such cases, it is recommended that a specialised
haemophilia centre be contacted. The risk of developing inhibitors is correlated to the extent of
exposure to factor VIII, the risk being highest within the first 20 exposure days, and to other genetic
and environmental factors. Rarely, inhibitors may develop after the first 100 exposure days. Cases of
recurrent inhibitor (low titre) have been observed after switching from one factor VIII product to
another in previously treated patients with more than 100 exposure days who have a history of
inhibitor development. Patients treated with coagulation factor VIII should be carefully monitored for
the development of inhibitors by appropriate clinical observations and laboratory tests (see section
4.8).
After reconstitution this medicinal product contains 0.45 mmol sodium (10 mg) per vial. To be taken
into consideration by patients on a controlled sodium diet.
24
No interaction studies have been performed with ADVATE.
Animal reproduction studies have not been conducted with factor VIII. Based on the rare occurrence
of haemophilia A in women, experience regarding the use of factor VIII during pregnancy and breast-
feeding is not available. Therefore, factor VIII should be used during pregnancy and lactation only if
clearly indicated.
ADVATE has no influence on the ability to drive and use machines.
As with any intravenous protein product allergic type hypersensitivity reactions are possible. (see
section 4.4).
During clinical studies with ADVATE, a total of 56 adverse drug reactions (ADRs) were reported in
27 of 234 unique treated patients. The ADRs that occurred in the highest number of patients were
development of factor VIII inhibitors (5 patients), all occurring in previously untreated patients who
had elevated risk of inhibitor development, headache (5 patients), fever and dizziness (3 patients
each). Of the 56 ADRs, none were reported in neonates, 16 were reported in 13/32 infants, 7 were
reported in 4/56 children, 8 were reported in 4/31 adolescents and 25 were reported in 14/94 adults.
5
Frequency categories are defined according to the following convention: very common (≥1/10),
common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare
(<1/10,000), not known (cannot be calculated from the available data). Within each frequency
grouping, undesirable effects are presented in order of decreasing seriousness.
The following table 2 provides the frequency of adverse drug reactions in clinical trials:
Table 2
Frequency of adverse drug reactions (ADRs) in clinical trials
MedDRA Standard
System Organ Class
Adverse reaction
Patients
Adverse
Experience
Rate
(% patients)
a
Frequency
Infections and
infestations
Influenza
1
0.43
Uncommon
Laryngitis
1
0.43
Uncommon
Blood and lymphatic
system disorders
Lymphangitis
1
0.43
Uncommon
Nervous system
disorders
Headache
5
2.14
Common
Dizziness
3
1.28
Common
Memory impairment
1
0.43
Uncommon
Tremor
1
0.43
Uncommon
Migraine
1
0.43
Uncommon
Dysgeusia
1
0.43
Uncommon
5
Neonates (age 0 – 1 month), infants (age 1 month – 2 years), children (age 2 - 12 years), adolescents (age 12 -
16 years) and adults (age over 16 years)
25




























Table 2
Frequency of adverse drug reactions (ADRs) in clinical trials
MedDRA Standard
Adverse reaction
Patients
Adverse
Frequency
System Organ Class
Experience
Rate
(% patients)
a
Eye disorders
Eye inflammation
1
0.43
Uncommon
Vascular disorders
Haematoma
1
0.43
Uncommon
Hot flush
1
0.43
Uncommon
Pallor
1
0.43
Uncommon
Respiratory, thoracic and
mediastinal disorders
Dyspnoea
1
0.43
Uncommon
Gastrointestinal
disorders
Diarrhoea
2
0.85
Uncommon
Abdominal pain upper
1
0.43
Uncommon
Nausea
1
0.43
Uncommon
Vomiting
1
0.43
Uncommon
Skin and subcutaneous
tissue disorders
Pruritus
2
0.85
Uncommon
Rash
2
0.85
Uncommon
Hyperhidrosis
1
0.43
Uncommon
Dermatitis diaper
1
0.43
Uncommon
General disorders and
administration site
conditions
Pyrexia
3
1.28
Common
Oedema peripheral
1
0.43
Uncommon
Chest pain
1
0.43
Uncommon
Chills
1
0.43
Uncommon
Feeling abnormal
1
0.43
Uncommon
Investigations
Anti-factor VIII antibody
positive
5
2.14
Common
Alanine
aminotransferase
increased
1
0.43
Uncommon
Coagulation factor VIII
level decreased
b
1
0.43
Uncommon
Haematocrit decreased
1
0.43
Uncommon
Laboratory test abnormal
1
0.43
Uncommon
Injury, poisoning and
procedural complications
Post procedural
complication
1
0.43
Uncommon
Post procedural
haemorrhage
1
0.43
Uncommon
Procedural site reaction
1
0.43
Uncommon
a) Percent of patients calculated based on total number of unique patients (234).
b) The unexpected decrease in coagulation factor VIII levels occurred in one patient during
continuous infusion of ADVATE following surgery (postoperative days 10-14). Haemostasis
was maintained at all times during this period and both plasma factor VIII levels and clearance
rates returned to appropriate levels by postoperative day 15. Factor VIII inhibitor assays
performed after completion of continuous infusion and at study termination were negative.
26































































Inhibitor Development
The immunogenicity of ADVATE was evaluated in previously treated patients. During clinical trials
haemophilia A (FVIII ≤ 2%), with previous exposure to factor VIII concentrates ≥ 150 days, one
patient developed a low-titre inhibitor (2.4 BU in the modified Bethesda assay) after 26 exposure days
to ADVATE. Follow-up inhibitor tests in this patient after withdrawal from the study were negative.
Also, in 53 paediatric patients under the age of 6, also diagnosed with severe to moderately severe
haemophilia A (FVIII ≤ 2%), with previous exposure to factor VIII concentrates ≥ 50 days, no FVIII
inhibitor was detected.
In previously untreated patients in an ongoing clinical study, 5 (20%) of 25 patients who received
ADVATE developed inhibitors to factor VIII. Of those, 4 were high-titre (≥ 5 BU) and 1 was low-titre
(< 5 BU). The frequency of FVIII inhibitors detected so far is within the expected and previously
observed range.
The patient’s immune response to trace amounts of contaminating proteins was analysed by examining
titres of antibodies to these proteins, laboratory parameters and reported adverse events. Of the 182
treated patients who were assessed for antibodies to Chinese hamster ovary (CHO) cell protein, 3
showed a statistically significant upward trend in titres by linear regression analysis and 4 displayed
sustained peaks or transient spikes. One patient had both a statistically significant upward trend and
displayed a sustained peak in anti-CHO protein antibody level, but no other signs or symptoms
indicative of an allergic or hypersensitivity response. Of the 182 treated patients who were assessed
for antibodies to murine IgG, 10 showed a statistically significant upward trend by linear regression
analysis and 2 displayed a sustained peak or transient spike. One patient had both a statistically
significant upward trend and displayed a sustained peak in anti-murine IgG antibody level. Four of
these patients reported isolated events of urticaria, pruritus, rash, and slightly elevated eosinophil
counts amongst numerous repeated exposures to the study product.
As with other intravenous products, allergic type hypersensitivity reactions, including
anaphylaxis/anaphylactoid reactions, have been reported with ADVATE (frequency not known).
No symptoms of overdose with recombinant coagulation factor VIII have been reported.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antihaemorrhagics, blood coagulation factor VIII. ATC code: B02BD02.
The factor VIII/von Willebrand Factor complex consists of two molecules (factor VIII and von
Willebrand factor) with different physiological functions. ADVATE contains recombinant coagulation
factor VIII (octocog alfa), a glycoprotein that is biologically equivalent to the factor VIII glycoprotein
found in human plasma.
Octocog alfa is a glycoprotein consisting of 2332 amino acids with an approximate molecular mass of
280 kD. When infused into a haemophilia patient, octocog alfa binds to endogenous von Willebrand
Factor in the patient’s circulation. Activated factor VIII acts as a Cofactor for activated Factor IX,
accelerating the conversion of Factor X to activated Factor X. Activated Factor X converts
prothrombin to thrombin. Thrombin then converts fibrinogen into fibrin and a clot can be formed.
Haemophilia A is a sex-linked hereditary disorder of blood coagulation due to decreased levels of
factor VIII activity and results in profuse bleeding into joints, muscles or internal organs, either
6
paediatric patients (age 0 –16 years) and adult patients (age over 16 years)
27
spontaneously or as a result of accidental or surgical trauma. The plasma levels of factor VIII are
increased by replacement therapy, thereby enabling a temporary correction of the factor VIII
deficiency and correction of the bleeding tendency.
5.2 Pharmacokinetic properties
All pharmacokinetic studies with ADVATE were conducted in previously treated patients with severe
to moderately severe haemophilia A (baseline factor VIII ≤ 2%). The analysis of plasma samples was
conducted in a central laboratory using a one-stage clotting assay. The pharmacokinetic parameters
derived from a crossover study of ADVATE in 100 previously treated patients ≥ 10 years of age are
listed in table 3 below.
Table 3
Summary of pharmacokinetic parameters for ADVATE in 100 patients with severe to
moderately severe haemophilia A (factor VIII ≤ 2%)
PK Parameter
Mean
SD
Median
Interquartile
Range
AUC
0-∞
(IU·h/dl)
1527
*
528
1549
668
t
1/2
(h)
11.8
3.1
11.1
2.8
Adjusted Recovery
(IU/dl
/
IU/kg)
2.38
*
0.50
2.42
0.68
C
max
(IU/dl)
120
*
26
119
38
Clearance (dl/kg·h)
0.037
0.030
0.033
0.0147
MRT (h)
15.2
4.8
14.1
4.2
V
SS
(dl/kg)
0.52
0.39
0.48
0.13
* Geometric mean
Data from single-dose administrations of ADVATE in 53 paediatric patients less than 6 years of age
were used to obtain this table.
Table 4
Summary of pharmacokinetic parameters for
ADVATE in 53 paediatric patients with severe to moderately
severe haemophilia A
PK Parameter
Mean
SD
AUC
0-∞
(IU·h/dl)
1195
*
430
t
1/2
(h)
9.7
1.9
Adjusted Recovery
(IU/dl
/
IU/kg)
1.84
*
0.42
C
max
(IU/dl)
93
*
22
Clearance (dl/kg·h)
0.044
0.014
MRT (h)
12.2
3.1
V
SS
(dl/kg)
0.51
0.12
*
Geometric Mean
Adjusted recovery and terminal half-life (
t1/2
) was approximately 20% lower in young children (less
than 6 years of age) than in adults, which may be due in part to the known higher plasma volume per
kilogram body weight in younger patients.
Pharmacokinetic data with ADVATE on previously untreated patients are currently not available.
28





























5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on studies of safety pharmacology, acute
toxicology, repeated dose toxicity, local toxicity and genotoxicity.
6.
6.1 List of excipients
Powder
Mannitol
Sodium chloride
Histidine
Trehalose
Calcium chloride
Trometamol
Polysorbate 80
Glutathione (reduced)
Solvent
Sterilised water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products or solvents.
6.3 Shelf life
Unopened powder vial
2 years.
After reconstitution
Chemical and physical in-use stability has been demonstrated for 3 hours at 25°C. From a
microbiological point of view, the product should be used immediately after reconstitution.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C). Do not freeze.
During the shelf life, the product may be kept at room temperature (up to 25°C) for a single period not
exceeding 6 months. The beginning of storage at room temperature should be recorded on the product
carton. The product may not be returned to refrigerated storage again.
Keep the vial in the outer carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
6.5 Nature and contents of container
Each pack contains a powder vial, a vial containing 5 ml solvent (both type I glass closed with
chlorobutyl rubber stoppers) and a device for reconstitution (BAXJECT II)
.
29
6.6 Special precautions for disposal and other handling
ADVATE is to be administered intravenously after reconstitution of the lyophilized product with the
provided sterilised water for injections. After reconstitution the solution should be clear, colourless
and free from foreign particles.
-
For reconstitution use only the sterilised water for injections and the reconstitution device
provided in the pack. For administration the use of a luer-lock syringe is required.
-
Use within three hours after reconstitution.
-
Dispose any unused product or waste material in accordance with local requirements.
-
Do not use if the BAXJECT II device, its sterile barrier system or its packaging is damaged or
shows any sign of deterioration.
Reconstitution
Aseptic Technique should be used
1.
If the product is still stored in a refrigerator, take both the ADVATE powder and solvent vials
from the refrigerator and let them reach room temperature (between 15°C and 25°C).
2.
Wash your hands thoroughly using soap and warm water.
3.
Cleanse stoppers with alcohol swabs. Place the vials on a flat clean surface.
5.
Open the package of BAXJECT II device by peeling away the paper lid without touching the
inside (Fig. a). Do not remove the device from the package. Do not use if the BAXJECT II
device, its sterile barrier system or its packaging is damaged or shows any sign of deterioration.
6.
Turn the package over and insert the clear plastic spike through the solvent stopper. Grip the
package at its edge and pull the package off BAXJECT II (Fig. b). Do not remove the blue cap
from the BAXJECT II device.
7.
For reconstitution only the water for injections and the reconstitution device provided in the
pack should be used. With BAXJECT II attached to the solvent vial, invert the system so that
the solvent vial is on top of the device. Insert the white plastic spike through the ADVATE
stopper. The vacuum will draw the solvent into the ADVATE vial (Fig. c).
8.
Swirl gently until all material is dissolved. Be sure that ADVATE is completely dissolved,
otherwise not all reconstituted solution will pass through the device filter. The product dissolves
rapidly (usually in less than 1 minute). After reconstitution the solution should be clear,
colourless and free from foreign particles.
Fig. a
Fig. b
Fig. c
Administration
Use Aseptic Technique
Parenteral medicinal products should be inspected for particulate matter prior to administration,
whenever solution and container permit. Only a clear and colourless solution should be used.
1.
Remove the blue cap from BAXJECT II. DO NOT DRAW AIR INTO THE SYRINGE.
Connect the syringe to BAXJECT II (Fig. d).
30
-
Do not refrigerate the preparation after reconstitution.
4.
Remove caps from powder and solvent vials.
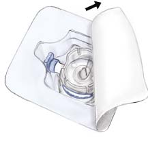



2.
Invert the system (the vial with the reconstituted solution has to be on top). Draw the
reconstituted solution into the syringe by pulling the plunger back slowly (Fig. e).
3.
Disconnect the syringe.
4.
Attach a butterfly needle to the syringe. Inject intravenously. The solution should be
administered slowly, at a rate as determined by the patient’s comfort level, not to exceed 10 ml
per minute. The pulse rate should be determined before and during administration of ADVATE.
Should a significant increase occur, reducing the rate of administration or temporarily
interrupting the injection usually allows the symptoms to disappear promptly (see sections 4.4
and 4.8).
Fig. d
Fig. e
7.
Baxter AG
Industriestrasse 67
A-1221 Vienna
Austria
8.
EU/1/03/271/003
9.
Date of first authorisation: 02 March 2004
Date of last renewal: 02 March 2009
Detailed information on this product is available on the website of the European Medicines Agency
(EMEA)
http://www.emea.europa.eu/
31
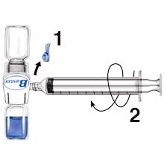

1.
Marketing Authorisation Holder
Baxter AG
Industriestrasse 67
A-1221 Vienna
145














