










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Rilonacept Regeneron 80 mg/ml powder and solvent for solution for injection.
2.
Each vial of powder contains 220 mg of rilonacept. After reconstitution, each ml of solution contains 80
mg rilonacept.
For a full list of excipients, see section 6.1.
Powder and solvent for solution for injection.
The powder is white to off-white.
The solvent is a clear colourless solution.
4.
Rilonacept Regeneron is indicated for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS)
with severe symptoms, including Familial Cold Autoinflammatory Syndrome (FCAS) and Muckle-Wells
Syndrome (MWS), in adults and children aged 12 years and older.
Treatment should be initiated and supervised by a specialist physician experienced in the diagnosis and
treatment of CAPS.
After proper training in the correct injection technique, patients may self-inject Rilonacept Regeneron if
their physician determines that it is appropriate and with medical follow-up as necessary.
Posology
Adults
Treatment in adults should be initiated with a loading dose of 320 mg. Dosing should be continued with a
once-weekly injection of 160 mg. Rilonacept Regeneron should not be given more often than once
weekly.
Paediatric population (12 to 17 years old)
Treatment should be initiated with a loading dose of 4.4 mg/kg, up to a maximum of 320 mg. Dosing
should be continued with a once-weekly injection of 2.2 mg/kg, up to a maximum of 160 mg (see Table
1). Dosing in children must be adjusted as the child grows. The patient or care giver should be advised to
speak to the treating physician before adjusting the dose. The experience in children is limited. In the
clinical program for CAPS, 8 adolescents aged 12-17 were treated for up to 18 months.
Paediatric population (up to 12 years old)
No data are available on the use of Rilonacept Regeneron in children with CAPS under 12 years of age,
therefore it is not recommended in this paediatric age group.
Elderly (65 years old or older)
Available data indicates that
dose modification is not required based on advanced age. However, clinical
experience in patients above 65 years is limited, therefore caution is recommended (see section 5.1).
Renal impairment
No dose adjustment is required in patients with mild, moderate, or severe renal impairment, or end stage
renal disease. However, clinical experience in such patients is limited.
2
Hepatic impairment
Rilonacept Regeneron has not been studied in patients with hepatic impairment.
Method of administration
Rilonacept Regeneron is for subcutaneous use only. It is not intended for intravenous or intramuscular
use.
For preparation and additional administration instructions, see section 6.6.
The adult loading dose should be administered as two 2 ml subcutaneous injections (320 mg of rilonacept
in total) given on the same day at different sites. The subsequent doses are administered as a 2 ml (160 mg
of rilonacept) subcutaneous injection once a week.
For paediatric patients, the dose is delivered as one or two (for loading dose) subcutaneous injections with
a maximum single-injection volume of 2 ml.
For convenience, the corresponding dose volume for weekly injection in paediatric patients is presented in
Table 1 below.
Table 1: Rilonacept Regeneron dose volume (after reconstitution) by body weight for paediatric patients
aged 12-17 years
Weight range (kg)
Dose volume (ml)
23.6 to 27.2
0.7
27.3 to 30.8
0.8
30.9 to 34.4
0.9
34.5 to 38.1
1
38.2 to 41.7
1.1
41.8 to 45.4
1.2
45.5 to 49.0
1.3
49.1 to 52.6
1.4
52.7 to 56.3
1.5
56.4 to 59.9
1.6
60.0 to 63.5
1.7
63.6 to 67.2
1.8
67.3 to 70.8
1.9
70.9 or greater
2
Hypersensitivity to rilonacept or to any of the excipients.
Active, severe infections (see section 4.4).
Serious infections
Interleukin-1 (IL-1) blockade may interfere with immune response to infections. Serious, life-threatening
infections have been reported uncommonly in patients taking Rilonacept Regeneron.
In an open-label extension study, one patient developed bacterial meningitis and died. Rilonacept
Regeneron should be discontinued if a patient develops a serious infection. Treatment should not be
initiated in patients with an active or chronic infection (see section 4.3) and physicians should exercise
caution when administering Rilonacept Regeneron to patients with a history of recurring infections or with
underlying conditions that may predispose them to infections.
Because Rilonacept Regeneron dampens an inflammatory response, vigilance in excluding underlying
infection in unwell patients is required
Tumour necrosis factor (TNF) inhibitors have been associated with an increased risk of reactivation of
latent tuberculosis (TB). It is unknown whether the use of IL-1 inhibitors like rilonacept increases the risk
of reactivation of TB or of opportunistic infections. Before starting treatment with Rilonacept Regeneron,
all patients should be evaluated for both active and inactive (latent) tuberculosis.
Combinations not recommended
3


















The combination of Rilonacept Regeneron with TNF inhibitors has not been evaluated in clinical studies.
An increased incidence of serious infections has been associated with administration of another IL-1
inhibitor, in combination with a TNF inhibitor.
Rilonacept Regeneron should not be used with TNF inhibitors because of increased risk of serious
infections
(see section 4.5).
The concomitant use of Rilonacept Regeneron with other IL-1 inhibitors is not recommended (see section
4.5).
Hypersensitivity
Although hypersensitivity reactions related to treatment with Rilonacept Regeneron were not seen in the
initial clinical program, if a hypersensitivity reaction occurs, administration should be discontinued and
appropriate therapy initiated.
The risk for severe hypersensitivity reactions, which is not uncommon for injectable proteins, cannot be
excluded (see section 4.3).
Immunogenicity
Antibodies directed against the receptor domains of rilonacept were detected by an ELISA assay in 35%
of patients (19 out of 55) treated for at least 6 weeks in the clinical study. There was no correlation of
antibody activity with either clinical efficacy or safety.
Neutropenia
Neutropenia (absolute neutrophil count [ANC] < 1.5 x 10
9
/l) has been observed commonly with another
medicinal product that inhibits IL-1 used in a patient population (rheumatoid arthritis) other than CAPS.
Neutropenia was observed commonly in patients with rheumatoid arthritis (not an approved use) who
were administered Rilonacept Regeneron subcutaneously in clinical studies. None of these patients had
serious infections associated with the neutropenia. Although neutropenia was observed uncommonly in
CAPS patients, the numbers studied are small. Treatment with Rilonacept Regeneron should not be
initiated in patients with neutropenia. It is recommended that neutrophil counts be assessed prior to
initiating treatment, after 1 to 2 months, and periodically thereafter while receiving Rilonacept Regeneron.
If a patient becomes neutropenic the ANC should be monitored closely and treatment discontinuation
should be considered.
Malignancies
The impact of treatment with Rilonacept Regeneron on the development of malignancies is not known.
However, treatment with immunosuppressants, including Rilonacept Regeneron, may result in an increase
in the risk of malignancies.
Vaccines
Live vaccines should not be given concurrently with Rilonacept Regeneron (see section 4.5). Prior to
initiation of Rilonacept Regeneron therapy, adult and paediatric patients should receive all recommended
vaccinations, as appropriate, including pneumococcal vaccine and inactivated influenza vaccine.
Lipid profile changes
Patients should be monitored for changes in their lipid profiles and provided with medical treatment if
warranted (see section 4.8).
Mutation in NLRP3 gene
All cases in the clinical trials had a confirmed mutation in the NLRP3 gene. The efficacy was not
evaluated in patients without a confirmed NLRP3 gene mutation.
No interaction studies have been performed.
The concomitant administration of Rilonacept Regeneron with any TNF inhibitor is not recommended
(see section 4.4), because an increased incidence of serious infections has been associated with
administration of another IL-1 blocker in combination with TNF inhibitors.
The concomitant administration of Rilonacept Regeneron with other IL-1 inhibitors has not been studied
and is therefore not recommended.
The formation of CYP450 enzymes is suppressed by increased levels of cytokines during chronic
inflammation. Thus it is expected that for a molecule that binds to IL-1, such as rilonacept, the formation
4
of CYP450 enzymes could be normalized. This is clinically relevant for CYP450 substrates with a narrow
therapeutic index, where the dose is individually adjusted (e.g. warfarin). Upon initiation of Rilonacept
Regeneron, in patients being treated with these types of medicinal products, therapeutic monitoring of the
effect or plasma levels should be performed and the individual dose of the medicinal product may need to
be adjusted as needed.
No data are available on either the effects of live vaccination or the secondary transmission of infection by
live vaccines in patients receiving Rilonacept Regeneron. Therefore, live vaccines should not be given
concurrently with Rilonacept Regeneron, unless the benefits clearly outweigh the risks. Should
vaccination with live vaccines be indicated after initiation of Rilonacept Regeneron treatment, the
recommendation is to wait for at least 6 weeks after the last Rilonacept Regeneron injection and before the
next one (see section 4.4).
Pregnancy
There are no adequate data from use of rilonacept in pregnant women. Reproductive toxicity studies have
been conducted in animals and have shown no effects on fertility or foetal morphology; however a study
in pregnant monkeys showed reduced levels of oestrogen (see section 5.3). The risk for the foetus/mother
is unknown. Women should use effective contraceptives during treatment with Rilonacept Regeneron and
for up to 6 weeks after the last dose. Women who are pregnant or who desire to become pregnant should
therefore only be treated after a thorough benefit-risk evaluation.
Lactation
It is unknown whether rilonacept is excreted in human or animal breast milk. A decision on whether to
continue/discontinue breast-feeding or to continue/discontinue therapy with Rilonacept Regeneron should
be made taking into account the benefit of breast-feeding to the child and the benefit of Rilonacept
Regeneron therapy to the woman.
The ability to drive and operate machines may be impaired by some symptoms associated with CAPS.
Patients who experience vertigo during Rilonacept Regeneron treatment should wait for this to resolve
completely before driving or operating machines.
The majority of the related adverse events in the clinical trials were classified as injection site reactions,
experienced by approximately 50% of the patients in the Phase 3 study. Reported ISRs were generally
mild to moderate in severity. No patients withdrew from the study due to ISRs.
ADRs to Rilonacept Regeneron reported during the Phase 2/3 program in a total of 109 patients, some
treated for longer than 2 years, are listed below using the following categories of frequency: very common
(≥1/10); common (≥ 1/100 to <1/10), uncommon (≥1/1000 to <1/100).
Due to the small patient population, an ADR reported in 2 or more patients is classified as
“common.”
5
Table 2: Adverse reactions with Rilonacept Regeneron in CAPS patients
MedDRA
System Organ Class
Adverse reaction
Frequency
General disorders and
administration site conditions
Injection site reactions,
including erythema, bruising,
pruritis, swelling,
inflammation, pain, dermatitis,
oedema, urticaria vesicles.
very common
Fatigue
common
Infections and infestations
Upper respiratory tract
infection; sinusitis
very common
Bronchitis; gastroenteritis;
viral infections; skin, eye and
ear infections; pneumonia
common
Bacterial meningitis
uncommon
Investigations
Eosinophil count increased
common
Nervous system disorders
Headache
very common
Dizziness
common
Vascular disorders
Hypertension, flushing
common
Ear and labyrinth disorders
Vertigo
common
Eye disorders
Iritis
uncommon
Psychiatric disorders
Anxiety, insomnia
common
Immune system disorders
Hypersensitivity
common
Infections and infestations
During Part A of the pivotal study (see section 5.1), the incidence of patients reporting infections and
considered by the investigator as related to treatment was greater with Rilonacept Regeneron (9%) than
with placebo (0%). In Part B, randomised withdrawal, the incidence of infections were similar in the
Rilonacept Regeneron (0%) and the placebo patients (4%). Part A of the trial was initiated in the winter
months, while Part B was predominantly performed in the summer months.
In placebo-controlled studies across a variety of patient populations encompassing 336 patients treated
with rilonacept and 165 treated with placebo, the incidence of infections was 6.8% and 3% (0.44 per
patient-exposure year and 0.19 per patient-exposure year), respectively, for rilonacept and placebo.
Serious infections
One patient in an open-label study of CAPS died after developing sinusitis and bacterial (
Streptococcus
pneumoniae
) meningitis.
In a study in patients with adult Still’s disease, one patient developed an infection in his elbow with
Mycobacterium intracellulare
after an intraarticular glucocorticoid injection and subsequent local
exposure to a suspected source of mycobacteria. In a study in patients with polymyalgia rheumatica, one
patient developed bronchitis and sinusitis, which resulted in hospitalization.
Blood and lymphatic system disorder
During the initial placebo-controlled portion of the pivotal trial, mean values increased for haemoglobin
and decreased for neutrophils and platelets in the patients treated with Rilonacept Regeneron. These
changes were not deemed as clinically significant and were potentially due to a decrease in the chronic
inflammatory state present in CAPS with an attendant decrease in acute-phase response.
General disorders and administration site conditions
In patients with CAPS, the most common and consistently reported adverse event associated with
treatment was injection-site reaction (ISR). The ISRs included erythema, swelling, pruritis, and bruising.
Most ISRs lasted for one to two days. In studies of patients with CAPS, no ISRs were assessed as severe,
and no patient discontinued study participation due to an ISR.
Immunogenicity
Antibodies directed against the receptor domains of rilonacept were detected by an ELISA assay in
patients with CAPS after treatment with Rilonacept Regeneron in clinical studies. Nineteen of 55 patients
(35%) who had received Rilonacept Regeneron for at least 6 weeks tested positive for treatment-emergent
binding antibodies on at least one occasion. Of the 19 patients, 7 tested positive at the last assessment
(Week 18 or 24 of the open-label extension period), and 5 patients tested positive for neutralising
antibodies on at least one occasion. There was no correlation of antibody activity and either clinical
efficacy or safety.
6


















The data reflect the percentage of patients whose test results were positive for antibodies to rilonacept in
specific assays, and are highly dependent on the sensitivity and specificity of the assays. The observed
incidence of antibody positivity in an assay may be influenced by several factors including assay
sensitivity and specificity, sample handling, concomitant medicinal products, and underlying disease. For
these reasons, comparison of the incidence of antibodies to rilonacept with the incidence of antibodies to
other products may be misleading.
Changes in lipid parameters
Cholesterol and lipid levels may be reduced in patients with chronic inflammation. Patients with CAPS
treated with Rilonacept Regeneron experienced mean increases from baseline for total cholesterol, HDL
cholesterol, LDL cholesterol, and triglycerides of 19 mg/dl, 2 mg/dl, 10 mg/dl, and 57 mg/dl respectively
after 6 weeks of open-label therapy. Physicians should monitor the lipid profiles of their patients (for
example after 2-3 months) and consider lipid-lowering therapies as needed based upon cardiovascular risk
factors and current guidelines.
No case of overdose has been reported. The maximum amount of product that can be safely administered
has not been determined.
Intravenous administration of Rilonacept Regeneron at doses of up to 2000 mg monthly in another patient
population for up to six months was generally well-tolerated. One patient in a study of osteoarthritis
developed transient neutropenia (absolute neutrophil count < 1 x 10
9
/l) after receiving a very large dose
(2000 mg). Maximum weekly doses of up to 320 mg have been administered subcutaneously for up to
approximately 2 years or more in a small number of patients with CAPS and up to 6 months in patients
with RA in clinical studies without evidence of dose-limiting toxicities.
In case of overdose, it is recommended that the patient be monitored for any signs or symptoms of adverse
reactions, and appropriate symptomatic treatment instituted immediately.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Interleukin inhibitors, ATC code: L04AC04.
This medicinal product has been authorised under “Exceptional Circumstances”. This means that due to
the rarity of the disease it has not been possible to obtain complete information on this medicinal product.
The EMA will review any new information, which may become available every year, and this SPC will be
updated as necessary.
Mechanism of action
Rilonacept is a dimeric fusion protein consisting of the ligand-binding domains of the extracellular
portions of the human type I interleukin-1 receptor (IL-1RI) and IL-1 receptor accessory protein
(IL-1RAcP) linked in-line to the Fc portion of human IgG1. Rilonacept binds to and blocks the activity of
the cytokine IL-1 and binds both IL-1
β
and IL-1
α
β
or IL-1
α
.
show a rapid response
to therapy with rilonacept, i.e. laboratory parameters such as C-reactive protein (CRP) and serum
amyloid A (SAA) levels, leukocytosis, and high platelet count rapidly returned to normal.
β
Clinical data
The safety and efficacy of rilonacept for the treatment of CAPS, including patients with FCAS, also
known as familial cold urticaria syndrome (FCUS), and MWS was demonstrated in a randomised,
double-blind, placebo-controlled study with two parts (A and B) conducted sequentially in the same
patients. The efficacy portion of the study included 47 patients, 44 of whom had a diagnosis of FCAS and
3 with a diagnosis of MWS. Twelve additional patients enrolled during the open label extension in which
efficacy data were collected, 8 adults with a diagnosis of FCAS and 4 adolescents (13-16 years old), 3
with FCAS and 1 with FCAS/MWS overlap. Four additional adolescents (12-17 years) all with a
diagnosis of FCAS subsequently enrolled in the open label extension where efficacy assessments were not
collected. The efficacy was not evaluated in patients without a confirmed NLRP3/CIAS1 gene mutation.
7
, which are the primary pro-inflammatory cytokines
implicated in many inflammatory diseases. Rilonacept also binds the endogenous IL-1 receptor antagonist
(IL-1ra) but with a lower affinity than IL-1
Pharmacodynamic effects
In clinical studies, CAPS patients who have uncontrolled over-production of IL-1
Part A was a 6-week, randomised, double-blind, placebo-controlled period to evaluate rilonacept at a dose
of 160 mg weekly after an initial loading dose of 320 mg. Immediately after Part A patients entered Part B
which consisted of a 9-week, patient-blind period during which all patients received rilonacept 160 mg
weekly, followed by a 9-week, double-blind, randomised withdrawal period in which patients were
randomly assigned to either remain on rilonacept 160 mg weekly or to receive placebo. Patients were then
given the option to enroll in a 24-week, open-label treatment extension phase during which all patients
were treated with rilonacept 160 mg weekly.
Using a daily diary questionnaire, patients rated the following five signs and symptoms of CAPS: joint
pain, rash, feeling of fever/chills, eye redness/pain, and fatigue, each on a scale of 0 (none, no severity) to
10 (very severe). The study evaluated the mean symptom score using the change from baseline to the end
of treatment.
The changes in mean symptom scores for the randomised parallel-group period (Part A) and the
randomised withdrawal period (Part B) of the study are shown in Table 3. Patients treated with rilonacept
experienced an 84% reduction in the mean symptom score in Part A compared to 13% for placebo-treated
patients (p< 0.0001). In Part B, mean symptom scores increased more in patients withdrawn to placebo
compared to patients who remained on rilonacept.
Improvement in key symptom scores was noted within one day of initiation of rilonacept therapy in most
patients. Patients treated with rilonacept experienced more improvement in each of the five components of
the composite endpoint than placebo-treated patients.
The mean number of symptomatic “flare” days (defined as a day in which the mean symptom score
reported on the patient diary was greater than 3) during the 21-day pre-treatment baseline period and the
on-treatment endpoint period, in Part A, decreased from 8.6 at baseline to 0.1 at endpoint for the group on
rilonacept, compared to a change from 6.2 to 5.0 for the placebo group (p<0.0001 vs. placebo).
A significantly higher proportion of patients in the rilonacept group compared to the placebo group
experienced improvement from baseline in the composite score by at least 30% (96% vs. 29% of patients),
by at least 50% (87% vs. 8%) and by at least 75% (70% vs. 0%) (p<0.0001).
In Part A and Part B, physician’s and patient’s global assessment of disease activity and patients’
assessment of the degree of limitation of their daily activities due to their disease were significantly
improved for patients treated with rilonacept compared with those on placebo.
Mean levels of C reactive protein (CRP) were significantly decreased versus baseline for the rilonacept-
treated patients, while there was no change for those on placebo. Rilonacept also led to a significant
decrease in serum amyloid A (SAA) versus baseline to levels within the normal range.
During the open-label extension, reductions in mean symptom scores, serum CRP, and serum SAA levels
were maintained for up to one year.
Table 3: Mean Symptom Scores in Adults (age 18 and o
lder)
Part A
Placebo
(n=24)
Rilonacept
(n=23)
Part B
Placebo
(n=23)
Rilonacept
(n=22)
Pre-treatment
Baseline Period
(Weeks -3 to 0)
2.4
3.1
Active Rilonacept
Baseline Period
(Weeks 13 to 15)
0.2
0.3
Endpoint Period
(Weeks 4 to 6)
2.1
0.5
Endpoint Period
(Weeks 22 to 24)
1.2
0.4
Mean Change from
Baseline to
Endpoint
-0.3
-2.5*
Mean Change from
Baseline to Endpoint
0.9
0.1**
p-value for within
group comparison
of change from
Baseline
NS
p< 0.0001
p-value for within
group comparison of
change from
Baseline
p< 0.0001 NS
*p< 0.0001, comparison of rilonacept vs. placebo
**p< 0.001, comparison of rilonacept vs. placebo,
NS = not significant
An assessment of efficacy with respect to age group and diagnosis was obtained by comparing KSS at the
end of the 24 week open label extension with KSS at baseline using time averaged daily mean scores. The
results for the adults who entered the study in Part A are provided separately from the results of the adults
8














who entered directly into the open label extension; the results for the four adolescents who entered directly
into the open label extension are provided individually.
Table 4: Key symptom scores by age and diagnosis following 24-week open label extension
Group
Age group
(range)
Diagnosis
Baseline
Mean KSS
Week 24
Mean KSS
Reduction
from Baseline
Adults who entered in
Part A
18 - <65
(24,63)
FCAS
n=31
2.9
0.7
75.9%
≥ 65
(67,78)
FCAS
n=10
2.4
0.4
77.3%
18 - <65
(22, 45)
MWS
n=3
3.3
0.2
90.5%
Adults who entered in
OLE
18 - <65
(18, 56)
FCAS
n=8
2.3
0.2
93.0%
Adolescents who entered
in OLE
13
FCAS
2.4
0.4
85.6%
15
FCAS
0.3
0.0
100%
16
FCAS
2.8
0.0
100%
13
FCAS/MWS
0.7
0.0
95.7%
5.2 Pharmacokinetic properties
Bioavailability of rilonacept after a subcutaneous injection is estimated to be approximately 50%.
The average trough levels of rilonacept were approximately 24 µg/ml at steady state following weekly
subcutaneous doses of 160 mg for up to 48 weeks in patients with CAPS. The steady state appeared to be
reached by 6 weeks.
Table 5: Rilonacept steady-state pharmacokineti
c properties
1
.
Parameter
Value
2
C
max
(mg/l)
31.5
AUC (day mg/l)
198
CL /F (l/day)
0.808
7.72
1
Based on population PK modelling
2
Derived values are presented.
Special populations
No pharmacokinetic data are available in patients with hepatic impairment. As with other large proteins
elimination of rilonacept is expected to be via proteolytic catabolism and target mediated clearance.
Consequently, impaired liver function is not expected to affect the pharmacokinetics of rilonacept in a
clinically significant way.
Results of a single-dose study in patients with end-stage renal disease (ESRD) indicate that the rate of
elimination of rilonacept was not decreased. Renal elimination of rilonacept is therefore considered to be a
minor pathway for clearance. No dose adjustment is needed in patients with renal impairment.
No study was conducted to evaluate the effect of age, gender, or body weight on rilonacept exposure.
Based on limited data obtained from the clinical study, steady-state trough concentrations were similar
between male and female patients. Age (26-78 years old) and body weight (50-120 kg) did not appear to
have a significant effect on trough rilonacept concentrations. The effect of race could not be assessed
because only Caucasian patients participated in the clinical studies in CAPS, reflecting the epidemiology
of the disease.
9
T
1/2
terminal (day)



























5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology and repeated-dose toxicity.
-estradiol levels were seen
in the treated groups, the significance of this finding is unknown. In a prenatal and postnatal reproductive
toxicology study in which mice were dosed subcutaneously, with a murine analogue of rilonacept at doses
of 20, 100 or 200 mg/kg three times per week (the highest dose is approximately 6-fold higher than the
160 mg maintenance dose based on body surface area), there were no treatment-related effects.
β
Genotoxicity or long term animal studies have not been performed to evaluate the mutagenic or
carcinogenic potential of rilonacept.
6.
6.1 List of excipients
Powder
Glycine
Arginine hydrochloride
Histidine
Histidine hydrochloride monohydrate
Polyethylene glycol 3350
Sucrose
Solvent
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3
Shelf life
Vial
2 years.
Diluted solution
From a microbiological safety point of view, the product should be used as soon as possible but within 3
hours of reconstitution, because it does not contain a preservative. If not used immediately, in-use storage
times and conditions prior to use are the responsibility of the user and should not be longer than 24 hours
at 2 to 8
°
C.
6.4
Special precautions for storage
Store in a refrigerator. Do not freeze.
Keep the vials in the outer carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
6.5
Nature and contents of container
Powder vial
20 ml clear type I glass vial with rubber stopper and lacquered flip-off aluminium seal containing 220 mg
rilonacept.
Solvent via
l
LDPE vials containing 5 ml water for injections
Each pack contains:
4 vials of powder for solution for injection
4 vials of solvent
8 disposable 3 ml syringes
10
Animal studies were conducted to assess reproductive toxicity. In mice, a murine analogue of rilonacept
had no effect on fertility. A study of embryo-foetal development was conducted with rilonacept in
monkeys at doses up to approximately 4 times the human dose. Decreases in
8 disposable 27 gauge, ½-inch needles
6.6 Special precautions for disposal and other handling
Instructions for reconstitution
Using aseptic technique, Rilonacept Regeneron powder should be reconstituted with 2.3 ml of solvent
(water for injections) prior to administration.
The 2.3 ml of solvent should be withdrawn from the solvent vial attached directly to a 3 ml syringe and
then injected into the powder vial for reconstitution using the 27 gauge, ½-inch needle (to obtain a final
reconstitution volume of 2.75 ml). The needle and syringe used for reconstitution with solvent should then
be discarded and should not be used for subcutaneous injections. After the addition of solvent, the vial
contents should be reconstituted by shaking the vial for approximately one minute and then allowing it to
sit for one minute. The resulting 80 mg/ml solution is sufficient to allow a withdrawable volume of up to 2
ml for subcutaneous administration.
The reconstituted solution is viscous, clear and colourless to pale yellow. Prior to injection, the
reconstituted solution should be carefully inspected for any discolouration or particulate matter. If there is
discolouration or particulate matter in the solution, the product must not be used.
Instructions for administration
Using aseptic technique, the recommended dose volume, up to 2 ml (160 mg) of the solution, should be
withdrawn with a new 27 gauge, ½-inch injection needle attached to a new 3 ml syringe for subcutaneous
injection.
Sites for subcutaneous injection, such as the abdomen, thigh, or upper arm, should be rotated. Injections
should never be made at sites that are bruised, red, tender, or hard.
The initial administration of Rilonacept Regeneron by a patient or caregiver should be under the guidance
of a trained healthcare professional. For subsequent self-administration by patients, appropriate instruction
in proper injection technique should be provided and ability to apply that technique ascertained.
Disposal
Each vial should be used for a single dose only. The vial should be discarded after withdrawal of the
solution.
Patients or their caregivers should be instructed on the appropriate procedure for disposal of the vials,
needles, and syringes.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Regeneron UK Limited
40 Bank Street
E14 5DS London
United Kingdom
8.
EU/1/09/582/001
9.
23/10/2009
Detailed information on this medicinal product is available on the website of the European Medicines
Agency (EMA)
http://www.ema.europa.eu
/.
11
ANNEX II
A.
MANUFACTURER OF THE BIOLOGICAL ACTIVE
SUBSTANCE ANDMANUFACTURING AUTHORISATION
HOLDER RESPONSIBLE FOR BATCH RELEASE
B.
CONDITIONS OF THE MARKETING AUTHORISATION
C.
12
A. MANUFACTURER OF THE BIOLOGICAL ACTIVE SUBSTANCE AND
MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturer of the biological active substance
Regeneron Pharmaceuticals, Inc.
81 Columbia Turnpike, Rensselaer,
New York 12144
USA
Name and address of the manufacturer responsible for batch release
Brecon Pharmaceuticals Ltd.
Pharos House
Wye Valley Business Park
Brecon Road, Hay-on-Wye
Hereford HR3 5PG
United Kingdom
B. CONDITIONS OF THE MARKETING AUTHORISATION
•
Medicinal product subject to restricted medical prescription (See Annex I: Summary of Product
Characteristics).
•
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND
EFFECTIVE USE OF THE MEDICINAL PRODUCT
The
Marketing Authorisation Holder (
MAH) shall ensure that, prior to launch, all physicians who are
expected to prescribe/use Rilonacept Regeneron are provided with a physician information pack
containing the following:
•
The Summary of Product Characteristics
•
Physician information
•
Patient Alert Card
The physician information should contain the following key messages:
•
The risk of serious infections, including opportunistic bacterial, viral and fungal infections in
patients treated with Rilonacept Regeneron;
•
The risk of acute injection-related reactions;
•
The need to instruct patients on proper techniques for self-administration when the patient is
willing and capable to do so, and guidance for Health Care Professionals on how to report
administration errors;
•
The identified or potential risk of immunogenicity that might lead to immune-mediated
symptoms;
•
The need for Health Care Professionals to perform an annual clinical assessment of patients
regarding a potential increased risk for the development of malignancies;
•
The need to measure neutrophil counts prior to initiating treatment, after 1 to 2 months and
periodically thereafter while receiving Rilonacept Regeneron as treatment with Rilonacept
Regeneron should not be initiated in patients with neutropenia;
•
The need to monitor patients for changes in their lipid profiles;
•
The unknown safety of Rilonacept Regeneron in pregnant and lactating women, thus the need for
physicians to discuss this risk with patients if they become or plan to become pregnant;
•
The proper patient management as regards the interaction with vaccination;
•
The possibility to include patients in the registry study to facilitate the collection of long term
efficacy and safety data;
•
The role and use of patient alert card.
13
•
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, as described in version 2 presented in
Module 1.8.1. of the Marketing Authorisation Application, is in place and functioning before and whilst
the product is on the market.
Risk Management Plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in the
Pharmacovigilance Plan, as agreed in version 2 of 13 March 2009 of the Risk Management Plan (RMP)
presented in Module 1.8.2. of the Marketing Authorisation Application and any subsequent updates of the
RMP agreed by the CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, the
updated RMP should be submitted at the same time as the next Periodic Safety Update Report (PSUR).
In addition, an updated RMP should be submitted
•
When new information is received that may impact on the current Safety Specification,
Pharmacovigilance Plan or risk minimisation activities
•
Within 60 days of an important (pharmacovigilance or risk minimisation) milestone being reached
•
At the request of the EMA
C. SPECIFIC OBLIGATIONS TO BE FULFILLED BY THE MARKETING
AUTHORISATION HOLDER
The Marketing Authorisation Holder shall complete the following programme of studies within the
specified time frame, the results of which shall form the basis of the annual reassessment of the
benefit/risk profile.
Area
Description
Due date
Clinical
SO1
The Applicant is requested to provide regular safety and efficacy
data from the Global Registry for both adults and children. The
fact that a limited number of paediatric patients were included in
the clinical studies combined with the lack of data on the effect of
long term IL-1ß suppression is a concern in view of the orphan
nature of the condition. The Applicant will need to propose a plan
to collect data from the registry on safety and efficacy in children;
particularly risk of infection and possible impairment of immune
reactions such as response to vaccinations and growth.
In addition the Applicant is requested to assess cases for whom
there is loss of efficacy to determine whether this is due to
changes over time in PK/PD or antibody development.
With PSUR
The Applicant is required to provide updates on the recruitment
rates and any intermediary results with the PSURs.
The patients should be included in the Registry until both
following conditions are met: 5 years recruitment period and 200
patients included.
Clinical
SO2
Further PK steady state exposure data (AUC, C
max
, C
min
during
steady state) especially for paediatric subjects is required. The
Applicant is requested to commit to a PK study in children.
01 July 2011
14







ANNEX III
LABELLING AND PACKAGE LEAFLET
15
A. LABELLING
16
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
OUTER CARTON
1.
Rilonacept Regeneron 80 mg/ml powder and solvent for solution for injection
rilonacept
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each vial of powder contains 220 mg rilonacept. After reconstitution, each ml of solution contains 80 mg
rilonacept.
3.
LIST OF EXCIPIENTS
Also contains: glycine, arginine hydrochloride, histidine, histidine hydrochloride monohydrate
polyethylene glycol 3350, sucrose, and water for injections.
4.
Powder and solvent for solution for injection.
Contains:
4 vials of powder containing 220 mg rilonacept
4 vials of 5 ml solvent
8 disposable 3 ml syringes
8 disposable 27-gauge, 1/2-inch needles
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Read the package leaflet before use.
Subcutaneous use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator. Do not freeze.
Keep the vials in the outer carton in order to protect from light.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Any unused product or waste material should be disposed of in accordance with local requirements.
Regeneron UK Limited
40 Bank Street
E14 5DS London
United Kingdom
17




























EU/1/09/582/001
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Justification for not including Braille accepted.
18














PARTICULARS TO APPEAR ON THE IMMEDIATE PACKAGING
POWDER VIAL LABEL
1.
Rilonacept Regeneron 80 mg/ml powder for solution for injection
rilonacept
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each vial contains 220 mg of rilonacept. When reconstituted, each ml solution contains 80 mg of
rilonacept.
3.
LIST OF EXCIPIENTS
Also contains: glycine, arginine hydrochloride, histidine, histidine hydrochloride monohydrate
polyethylene glycol 3350, sucrose
4.
Powder for solution for injection.
220 mg rilonacept
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Read the package leaflet before use.
Subcutaneous use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator. Do not freeze.
Keep the vial in the outer carton in order to protect from light.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Regeneron UK Limited
40 Bank Street
E14 5DS London
United Kingdom
EU/1/09/582/001
13. BATCH NUMBER
Lot
19
































14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Justification for not including Braille accepted.
20










MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
SOLVENT LABEL
1.
Solvent for Rilonacept Regeneron
2.
METHOD OF ADMINISTRATION
Read the package leaflet before use.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUMEN OR BY UNIT
5 ml
6.
OTHER
Water for injections.
21
















B. PACKAGE LEAFLET
22
PACKAGE LEAFLET: INFORMATION FOR THE USER
Rilonacept Regeneron 80 mg/ml powder and solvent for solution for injection
rilonacept
Read all of this leaflet carefully before you start using this medicine.
-
Keep this leaflet. You may need to read it again.
-
If you have further questions, ask your doctor or pharmacist.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even if their
symptoms are the same as yours.
-
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet, please
tell your doctor or pharmacist.
In this leaflet:
1. What Rilonacept Regeneron is and what it is used for
2. Before you use Rilonacept Regeneron
3. How to use Rilonacept Regeneron
4. Possible side effects
5. How to store Rilonacept Regeneron
6. Further information
1.
WHAT Rilonacept Regeneron IS AND WHAT IT IS USED FOR
Rilonacept Regeneron is used to treat adults and adolescents aged 12 years and older with severe
symptoms of Familial Cold Auto-inflammatory Syndrome (FCAS) and Muckle-Wells syndrome (MWS).
Rilonacept Regeneron can help reduce inflammation and the signs and symptoms of your disease, such as
rash, joint pain, fever, and fatigue.
Rilonacept Regeneron belongs to a group of medicines called interleukin inhibitors. Rilonacept Regeneron
blocks the activity of substances including interleukin-1 beta (IL-1 beta). In patients with CAPS, the body
produces excessive amounts of IL-1 beta. This may lead to symptoms such as fever, headache, fatigue,
skin rash, or painful joints and muscles. By blocking the activity of IL-1 beta, Rilonacept Regeneron leads
to an improvement in these symptoms.
If you have any questions about how Rilonacept Regeneron works or why this medicine has been
prescribed for you, ask your doctor.
2.
BEFORE YOU USE RILONACEPT REGENERON
Do not use Rilonacept Regeneron
•
if you are allergic (hypersensitive) to rilonacept or to any of the other ingredients of Rilonacept
Regeneron (listed in section 6, ‘What Rilonacept Regeneron contains’);
•
if you have an active, severe infection.
Take special care with Rilonacept Regeneron
You should tell your doctor if you have:
•
an infection;
•
tuberculosis or you have been in close contact with someone who has had tuberculosis;
•
a history of infections that keep coming back;
•
been scheduled to receive any vaccines.
•
signs of an allergic reaction such as difficulty breathing, nausea, dizziness, skin rash, palpitations,
or low blood pressure.
Rilonacept Regeneron is not recommended for children younger than 12 years of age.
Using other medicines
Please tell your doctor or pharmacist if you are using or have recently used any other medicines, including
medicines obtained without a prescription.
In particular, you should tell you doctor if you are using medicines containing any of the following active
substances:
•
Tumour Necrosis Factor blockers.
23
•
Other medicines that block interleukin 1.
•
Any other medicines for chronic disorders, as Rilonacept Regeneron can affect how the liver
processes some medicines, such as warfarin. Your doctor may need to perform some tests and adjust
the dose of such medicines.
Pregnancy and breast-feeding
−
You are advised to avoid becoming pregnant and must use adequate contraception while using
Rilonacept Regeneron and for up to six weeks after the last Rilonacept Regeneron treatment. It is
important to tell your doctor if you are pregnant, if you think you may be pregnant or if you plan to
get pregnant. Your doctor will discuss with you the potential risk of taking Rilonacept Regeneron
during pregnancy.
−
It is not known whether Rilonacept Regeneron passes into human milk. Your doctor will discuss
with you the potential risks of taking Rilonacept Regeneron before breast-feeding.
Rilonacept Regeneron has not been tested in pregnant women.
Rilonacept Regeneron should not be used during pregnancy unless clearly necessary.
The safety of Rilonacept Regeneron in breast-feeding women is unknown. If you are breast-feeding you
should ask your doctor or pharmacist before using Rilonacept Regeneron.
Driving and using machines
Some symptoms associated with CAPS or with Rilonacept Regeneron treatment, such as a spinning
sensation (known as vertigo), may affect your ability to drive or use machines. If you feel a spinning
sensation, do not drive or operate any tools or machines until you are feeling normal again.
Ask your doctor, nurse or pharmacist for advice before taking any medicine.
3.
HOW TO USE RILONACEPT REGENERON
Always use Rilonacept Regeneron exactly as your doctor has told you. You should check with your doctor
or pharmacist if you are not sure.
Rilonacept Regeneron is intended for subcutaneous use. This means that it is injected through a short
needle into the fatty tissue just under the skin.
Adults
(
including the elderly)
The initial dose is 320 mg (corresponding to 2 injections of 2 ml solution each), given on the same day at
2 different sites, followed by a maintenance dose of 160 mg (1 injection of 2 ml) injected once weekly
Adolescents (aged 12 to 17 years old)
The prescribed dose will depend on the body weight of the patient. The initial dose is 4.4 mg/kg body
weight, up to 320 mg, delivered as one or two injections. This is followed by a maintenance dose of
2.2 mg/kg, up to 160 mg, once weekly on the same day of the week. In both cases your doctor will
calculate the corresponding volume to inject. The dose of Rilonacept Regeneron may need to be adjusted
as the child grows. Talk with your doctor before making any dose adjustments.
How to inject Rilonacept Regeneron
Rilonacept Regeneron is injected under the skin (subcutaneously). The first injection of Rilonacept
Regeneron should be given under the supervision of a trained healthcare professional. You or your
caregiver will receive adequate training on how to reconstitute the powder (dissolve to make a solution),
prepare the dose, and administer the injection. Detailed instructions are also provided at the end of this
leaflet.
If you use more Rilonacept Regeneron than you should
There are no reported overdoses in patients. In case of a possible overdose you should contact your doctor
immediately.
If you forget to use Rilonacept Regeneron
If you miss a dose of Rilonacept Regeneron and remember within a few days, inject it as soon as you
remember. The next dose should be taken at the next regularly scheduled time. Do not take a double dose
to make up for the forgotten dose.
24
4
.
POSSIBLE SIDE EFFECTS
Like all medicines, Rilonacept Regeneron
can cause side effects, although not everybody gets them.
You must tell your doctor immediately if any of the following serious side effects occur while you are
using Rilonacept Regeneron:
•
Serious infections.
You should talk to your doctor immediately if you develop symptoms of an infection
while being treated with Rilonacept Regeneron, such as
prolonged fever (i.e. fever lasting longer than 3 days) or any other symptoms possibly related to an
infection, such as prolonged cough, prolonged headache or localised redness, warmth or swelling of your
skin.
You must stop treatment with Rilonacept Regeneron if you develop a severe infection.
•
Allergic reactions.
If you develop signs of an allergic (hypersensitivity) reaction during treatment with
Rilonacept Regeneron (for example, rash, swollen face, trouble breathing), tell your doctor immediately.
Side effects may occur with certain frequencies, which are defined as follows:
very common: affects 1 or more user in 10
common: affects 1 to up to 10 users in 100
uncommon: affects 1 to up to10 users in 1,000
rare: affects 1 to up to 10 users in 10,000
very rare: affects less than 1 user in 10,000
not known: frequency cannot be estimated from the available data.
Very common side effects
Injection-site reactions. These include redness, swelling, itching, and bruising at the injection site. Most
injection-site reactions are mild and will go away in one to two days.
Upper respiratory infection
Sinus infection
Headache
Common side effects
Viral infection
Bronchitis
Skin, eye, or ear infection
Fatigue
Increased blood pressure
Pneumonia
Stomach/Intestinal infection
Dizziness
Flushing
Allergic reaction
Anxiety
Insomnia
Uncommon side effects
Meningitis
Inflammation of the eye (iritis)
Changes in your cholesterol levels or in your blood counts may also occur. These will be monitored by
your doctor.
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet, please tell
your doctor or pharmacist.
Keep out of the reach and sight of children.
HOW TO STORE RILONACEPT REGENERON
Do not use Rilonacept Regeneron after the expiry date which is stated on the carton and vial label after
EXP. The expiry date refers to the last day of that month.
Store in a refrigerator. Do not freeze.
25
5.
Keep the vials in the outer carton in order to protect from light.
After preparing the Rilonacept Regeneron solution, it is best if used immediately because it does not
contain a preservative. If necessary, the product may be kept at room temperature, but should be used
within 3 hours of reconstitution
The reconstituted solution is viscous, clear and colourless to pale yellow. Prior to injection, the
reconstituted solution should be carefully inspected for any discolouration or particulate matter. If there is
discolouration or particulate matter in the solution, the product must not be used.
Medicines should not be disposed of via wastewater or household waste. Ask pharmacist how to dispose
of medicines no longer required. These measures will help to protect the environment.
6.
FURTHER INFORMATION
What Rilonacept Regeneron contains
- The active substance is rilonacept. Each vial of powder contains 220 mg rilonacept. After
reconstitution, each ml of solution contains 80 mg rilonacept.
- The other ingredients in the powder are glycine, arginine hydrochloride, histidine, histidine
hydrochloride monohydrate, polyethylene glycol 3350 and sucrose. The solvent is water for injections.
What Rilonacept Regeneron looks like and contents of the pack
Rilonacept Regeneron is provided as a powder and solvent for solution for injection. The powder is white
to off-white and the solvent is a colourless liquid.
Powder vial: 20 ml clear type 1 glass vial with rubber stopper and lacquered flip-off aluminum seal
containing 220 mg rilonacept.
Solvent: 5 ml transparent plastic (LDPE) vial containing 5 ml water for injections.
Each pack contains:
4 vials of powder for solution for injection
4 vials of solvent
8 disposable, 3 ml syringes
8 disposable 27 gauge, ½-inch needles
Marketing Authorisation Holder Regeneron UK Limited
40 Bank Street
E14 5DS London
United Kingdom
Manufacturer
Brecon Pharmaceuticals Ltd
Wye Valley Business Park
Hay-on-Wye
HR3 5PG Hereford
United Kingdom
This leaflet was last approved in
Detailed information on this medicine is available on the European Medicines Agency (EMA) web site:
26
Instructions for use
STEP 1: Setting up for an injection
1.
Wash your hands well with soap and water, and dry with a clean towel.
2.
Put the following items on a clean table, or other flat surface, for each injection (see Figure 1):
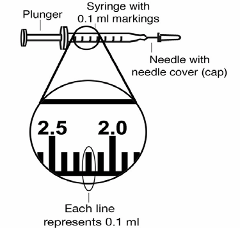
• 2 sterile, 3-ml disposable syringes with markings at each 0.1 ml (see Figure 2):
• one needed for dissolving (reconstituting) Rilonacept Regeneron
• one needed for injection
• 2 sterile disposable needles (27-gauge, ½-inch)
• one needed for dissolving (reconstituting) Rilonacept Regeneron
• one needed for injection
• 1 vial of Rilonacept Regeneron powder for solution for injection
• 1 vial of solvent (water for injections)
You will also need (not provided in the prescription pack):
• 2 alcohol wipes
• 1 2x2 gauze pad
• 1 puncture-resistant container for disposal of used needles, syringes and vials
Ask your pharmacist for these supplies.
Figure 1
Figure 2
• Do not touch the needles or the rubber stopper on the Rilonacept Regeneron vial with your hands. If you
do touch the rubber stopper, clean it with a fresh alcohol wipe.
• If you touch a needle or the needle touches any surface, throw away the entire syringe into the puncture
resistant container and start over with a new syringe.
• Do not reuse needles or syringes.
• To protect yourself and others from possible needle sticks, it is very important to throw away every
syringe, with the needle attached, in the puncture-proof container right after use.
Do not try to recap the
needle.
STEP 2: Preparing the Rilonacept Regeneron vial
1.
Check the expiration date on the carton of Rilonacept Regeneron. Do not use Rilonacept Regeneron
after the expiry date which is stated on the vial label and carton after EXP. The expiry date refers to
the last day of that month.
27
2.
Remove the protective plastic cap from the Rilonacept Regeneron vial.
3.
Clean the top of the Rilonacept Regeneron vial with an alcohol wipe, wiping in one direction
around the top
4.
Set the vial aside.
STEP 3: Preparing the solvent vial
1.
Snap off the plastic tab on the top of the solvent (water for injections) vial.
2. Open the wrapper that contains a 27-gauge needle by pulling apart the tabs. Place the capped needle
on a clean surface. Open the wrapper that contains the syringe by pulling apart the tabs.
3.
Attach the exposed top of the solvent vial to the top of a syringe by twisting the syringe onto the
solvent vial (see Figure3).
Figure 3
4.
Hold the vial in one hand and the syringe in the other hand and carefully turn the vial upside down.
Hold the syringe at eye level.
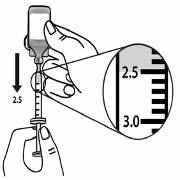
5.
Slowly pull back on the syringe plunger to the 2.5 ml line on the syringe (see Figure 4).
Figure 4
28
6.
Remove the vial from the syringe. Hold the barrel of the syringe with one hand and twist the 27-gauge
needle onto the tip of the syringe with the other hand until it fits snugly (see Figure 5).
Figure 5
7.
Turn the syringe so that the needle is facing straight up. Remove the needle cap. Gently tap the syringe
until air bubbles rise to the top of the syringe (see Figure 6).
Figure 6
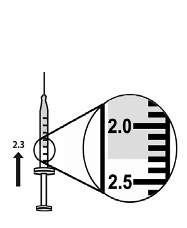
8. With the syringe and needle pointed upward push the syringe plunger to expel any excess solvent until
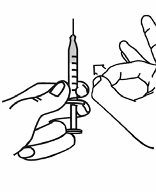
the 2.3 ml line is matched (see Figure 7).
Figure 7
29

STEP 4: Dissolving (reconstituting) Rilonacept Regeneron
1.
With one hand, hold the Rilonacept Regeneron vial on a firm surface.
2.
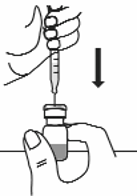
With the other hand, take the syringe with the solvent and slowly insert the needle straight down
through the centre of the rubber stopper of the Rilonacept Regeneron vial. Direct the water stream
to gently go down the side of the vial into the powder (see Figure 8).
Figure 8
3.
Push the plunger in all the way to inject the solvent into the vial.
4.
Remove the syringe and needle from the stopper and throw away the needle, syringe, and solvent
vial in the puncture-resistant container. Do not try to put the needle cover back on the needle.
5.
Hold the vial containing the mixture of powder and solvent (water for injections) sideways (not
upright) with your thumb and a finger at the top and bottom of the vial, and quickly shake the vial
back and forth (side-to-side) for about 1 minute.
6.
Put the vial back on the table and let the vial sit for about 1 minute.
7.
Look at the vial for any particles or clumps of powder which have not dissolved.
8.
If the powder has not completely dissolved, shake the vial quickly back and forth for 30 seconds
more. Let the vial sit for about 1 minute.
9.
Repeat step 8 until the powder is completely dissolved and the solution is clear.
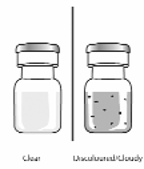
10. The dissolved Rilonacept Regeneron should be a thick, clear liquid, colourless to pale yellow in
colour. Do not use the solution if it is discoloured or cloudy, or if small particles are in it (see
Figure 9).
NOTE: Contact your pharmacy to report any dissolved Rilonacept Regeneron that is discoloured or
contains particles.
Figure 9
30
11. After preparing the solution, it is best if used immediately. If necessary, the product may be kept at
room temperature (20 to 25
C), but should be used within 3 hours of reconstitution. Keep
Rilonacept Regeneron away from light.
STEP 5: Preparing the injection
1.
Hold the vial with the solution on a firm surface and wipe the top of the powder vial with a new
alcohol wipe.
2.
Take a new sterile, disposable needle and attach securely to a new syringe without removing the
needle cover.
3.
The amount of air you draw into the syringe should equal the volume of the solution that your
doctor has prescribed for you to inject.
4.
To draw air into the syringe, hold the syringe at eye level. Do not remove the needle cover. Pull
back the plunger on the syringe to the mark that is equal to the volume of the solution that your
doctor has prescribed for you to inject (see Figure 10).
Figure 10
5.
Remove the needle cover and be careful not to touch the needle. Keep the vial on a flat surface and
slowly insert the needle straight down through the stopper. Push the plunger down and inject all the
air into the vial (see Figure 11).
Figure 11
6.
Hold the vial in one hand and the syringe in the other hand and carefully turn the vial upside down
so that the needle is pointing straight up. Hold the vial at eye level.
31
°

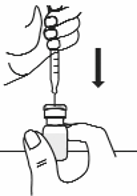
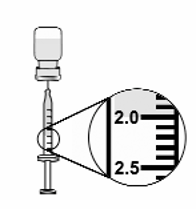
7.
Keep the tip of the needle in the liquid and slowly pull back on the plunger to the mark on the
syringe that matches the amount of medicine prescribed by your doctor (see Figure 12).
Figure 12
8.
Keep the vial upside down with the needle straight up, and gently tap the syringe until any air
bubbles rise to the top of the syringe (see Figure 13). It is important to remove air bubbles so that
you withdraw up the correct amount of medicine from the vial.
Figure 13
9.
To remove the air bubbles, slowly and gently push in the plunger so only the air is pushed through
the needle.
10. Check to make sure that you have the amount of medicine prescribed by your doctor in the syringe.
11. Throw away the vial in the puncture resistant container even if there is some medicine left in the
vial. Do not use any vial of Rilonacept Regeneron more than one time.
12. Hold the syringe and needle in your hand ready for injecting. Do not touch the needle with your
hands or allow it to come into contact with any surfaces. Proceed with the injection as described in
Step 6 below.
STEP 6: Giving the injection
1.
Rilonacept Regeneron is given by subcutaneous injection, an injection that is given into the tissue
directly below the layers of skin. It is not meant to go into any muscle, vein, or artery.
You should change (rotate) the sites and inject in a different place each time in order to keep your
skin healthy.
Rotating injection sites helps to prevent irritation and allows the medicine to be better absorbed.
Ask your doctor any questions that you have about rotating injection sites.
• Do not inject into skin that is tender, red, or hard. If an area is tender or feels hardened,
choose another site for injection until the tenderness or “hardening” goes away.
32
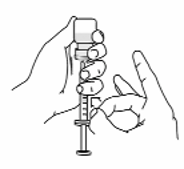
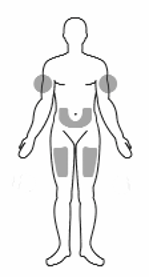
• Tell your doctor about any skin reactions including redness, swelling, or hardening of the
skin.
• Areas where you may inject Rilonacept Regeneron include the left and right sides of the
abdomen, and left and right thighs. If someone else is giving the injection, the upper left and
right arms may also be used for injection (see Figure 14):
(Do not inject within a 2-inch area around the navel)
Figure 14
2.
Choose the area for the injection. Clean the area in a circular motion with a new alcohol wipe.
Begin at the centre of the site and move outward. Let the alcohol air dry completely.
3.
Hold the syringe in one hand like you would hold a pencil.
4.
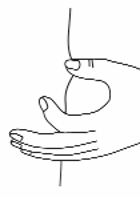
With the other hand gently pinch a fold of skin at the cleaned site for injection (see Figure 15).
Figure 15
33
5.
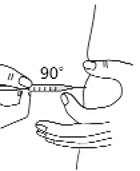
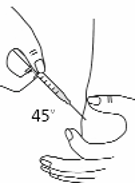
Use a quick “dart like” motion to insert the needle straight into the skin (90° angle) (see Figure 16).
Do not push down on the plunger while inserting the needle into the skin. For small children or
persons with little fat under the skin, you may need to hold the syringe and needle at a 45° angle
(see Figure 16).
Figure 16
6.
After the needle is completely in the skin, let go of the skin that you are pinching.
7.
With your free hand hold the syringe near its base. Gently pull back the plunger. If blood comes
into the syringe, the needle has entered a blood vessel. Remove the needle, discard the syringe and
needle. Start over with ‘STEP 1: Setting up for an injection’ using new supplies.
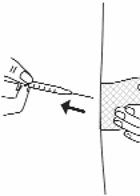
8. If no blood appears, inject all the medicine in the syringe at a slow, steady rate, pushing the plunger
all the way down. It may take up to 30 seconds to inject the entire dose.
9. Pull the needle out of the skin, and hold a piece of sterile gauze over the injection site for several
seconds (see Figure 17).
Figure 17
10. Do not replace the needle cover. Throw away the vials, used syringes and needles in the puncture-
resistant container. Do not recycle the container. Do not throw away vials, needles, or syringes in
the household rubbish.
11. Keep the puncture-resistant container out of reach of children. When the container is about two-
thirds full, dispose of it as instructed by your doctor or pharmacist.
12. Used alcohol wipes can be thrown away in the household rubbish.
34
-----------------------------------------------------------------------------------------------------------------------------
The following information is intended for medical or healthcare professionals only:
Indication
Rilonacept Regeneron is indicated for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS)
with severe symptoms, including Familial Cold Autoinflammatory Syndrome (FCAS) and Muckle-Wells
Syndrome (MWS), in adults and children aged 12 years and older.
Posology
Adults
Treatment in adults should be initiated with a loading dose of 320 mg. Dosing should be continued with a
once-weekly injection of 160 mg. Rilonacept Regeneron should not be given more often than once
weekly.
Paediatric population (12 to 17 years old)
Treatment should be initiated with a loading dose of 4.4 mg/kg, up to a maximum of 320 mg. Dosing
should be continued with a once-weekly injection of 2.2 mg/kg, up to a maximum of 160 mg (see Table
1). Dosing in children must be adjusted as the child grows. The patient or care giver should be advised to
speak to the treating physician before adjusting the dose. The experience in children is limited. In the
clinical program for CAPS, 8 adolescents aged 12-17 were treated for up to 18 months.
Paediatric population (up to 12 years old)
No data are available on the use of Rilonacept Regeneron in children with CAPS under 12 years of age,
therefore it is not recommended in this paediatric age group.
Elderly (65 years old or older)
Available data indicates that
dose modification is not required based on advanced age. However, clinical
experience in patients above 65 years is limited, therefore caution is recommended.
Renal impairment
No dose adjustment is required in patients with mild, moderate or severe renal impairment, or end stage
renal disease. However, clinical experience in such patients is limited.
Hepatic impairment
Rilonacept Regeneron has not been studied in patients with hepatic impairment.
Method of administration
Rilonacept Regeneron is for subcutaneous use only. It is not intended for intravenous or intramuscular
use.
The adult dose loading dose should be administered as two 2 ml subcutaneous injections (320 mg of
rilonacept in total) given on the same day at different sites. The subsequent doses are administered as a 2
ml (160 mg of rilonacept) subcutaneous injection once a week.
For paediatric patients, the dose is delivered as one or two (for loading dose) subcutaneous injections with
a maximum single-injection volume of 2 ml.
For convenience, the corresponding dose volume for weekly injection in paediatric patients is presented in
Table 1 below.
35
Table 1: Rilonacept Regeneron dose volume (after reconstitution) by body weight for paediatric patients,
aged 12-17 years
Weight range (kg)
Dose volume (ml)
23.6 to 27.2
0.7
27.3 to 30.8
0.8
30.9 to 34.4
0.9
34.5 to 38.1
1
38.2 to 41.7
1.1
41.8 to 45.4
1.2
45.5 to 49.0
1.3
49.1 to 52.6
1.4
52.7 to 56.3
1.5
56.4 to 59.9
1.6
60.0 to 63.5
1.7
63.6 to 67.2
1.8
67.3 to 70.8
1.9
70.9 or greater
2
Special precautions for storage
Store in a refrigerator. Do not freeze.
Keep the vials in the outer carton in order to protect from light.
After reconstitution, if necessary the product may be kept at room temperature, but should be used within
three hours of reconstitution because it does not contain a preservative.
Reconstitution and administration instructions
Instructions for reconstitution
Using aseptic technique, Rilonacept Regeneron powder should be reconstituted with 2.3 ml solvent (water
for injections) prior to administration.
The 2.3 ml of solvent should be withdrawn from the solvent vial attached directly to a 3-ml syringe and
then injected into the powder vial for reconstitution with 27-gauge, ½-inch needle (to obtain a final
reconstitution volume of 2.75ml). The needle and syringe used for reconstitution with solvent should then
be discarded and should not be used for subcutaneous injections. After the addition of solvent, the vial
contents should be reconstituted by shaking the vial for approximately one minute and then allowing it to
sit for one minute. The resulting 80 mg/ml solution is sufficient to allow a withdrawable volume of up to 2
ml for subcutaneous administration.
The reconstituted solution is viscous, clear and colourless to pale yellow. Prior to injection, the
reconstituted solution should be carefully inspected for any discolouration or particulate matter. If there is
discolouration or particulate matter in the solution, the product must not be used.
Instructions for administration
Using aseptic technique, the recommended dose volume, up to 2 ml (160 mg) of the solution should be
withdrawn with a new 27-gauge, ½-inch injection needle attached to a new 3-ml syringe for subcutaneous
injection.
Sites for subcutaneous injection, such as the abdomen, thigh, or upper arm, should be rotated. Injections
should never be made at sites that are bruised, red, tender, or hard.
The initial administration of Rilonacept Regeneron by a patient or caregiver should be under the guidance
of a trained healthcare professional. For subsequent self-administration by patients, appropriate instruction
in proper injection technique should be provided and ability to apply that technique ascertained.
Disposal
Each vial should be used for a single dose only. The vial should be discarded after withdrawal of the
solution.
Patients or their caregivers should be instructed on the appropriate procedure for disposal of the vials,
needles, and syringes.
36


















Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/arcalyst.html
Copyright © 1995-2021 ITA all rights reserved.