










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Arixtra 1.5 mg/0.3 ml solution for injection, pre-filled syringe.
2.
Each pre-filled syringe (0.3 ml) contains 1.5 mg of fondaparinux sodium.
Excipient(s): Contains less than 1 mmol of sodium (23 mg) per dose, and therefore is essentially
sodium free.
For a full list of excipients, see section 6.1.
3.
Solution for injection.
The solution is a clear and colourless liquid.
4.
Prevention of Venous Thromboembolic Events (VTE) in adults undergoing major orthopaedic surgery
of the lower limbs such as hip fracture, major knee surgery or hip replacement surgery.
Prevention of Venous Thromboembolic Events (VTE) in adults undergoing abdominal surgery who
are judged to be at high risk of thromboembolic complications, such as patients undergoing abdominal
cancer surgery (see section 5.1).
Prevention of Venous Thromboembolic Events (VTE) in adult medical patients who are judged to be
at high risk for VTE and who are immobilised due to acute illness such as cardiac insufficiency and/or
acute respiratory disorders, and/or acute infectious or inflammatory disease.
Treatment of adults with acute symptomatic spontaneous superficial-vein thrombosis of the lower
limbs without concomitant deep-vein thrombosis (see sections 4.2 and 5.1).
Posology
Patients undergoing major orthopaedic or abdominal surgery
The recommended dose of fondaparinux
is 2.5 mg once daily administered post-operatively by
subcutaneous injection.
The initial dose should be given
6 hours following surgical closure provided that haemostasis has been
established.
Treatment should be continued until the risk of venous thrombo-embolism has diminished, usually
until the patient is ambulant, at least 5 to 9 days after surgery. Experience shows that in patients
undergoing hip fracture surgery, the risk of VTE continues beyond 9 days after surgery. In these
patients the use of prolonged prophylaxis with fondaparinux should be considered for up to an
additional 24 days (see section 5.1).
2
Medical patients who are at high risk for thromboembolic complications based on an individual risk
assessment
The recommended dose of fondaparinux is 2.5 mg once daily administered by subcutaneous injection.
A treatment duration of 6-14 days has been clinically studied in medical patients (see section 5.1).
Treatment of superficial-vein thrombosis
The recommended dose of fondaparinux is 2.5 mg once daily, administered by subcutaneous injection.
Patients eligible for fondaparinux 2.5 mg treatment should have acute, symptomatic, isolated,
spontaneous superficial-vein thrombosis of the lower limbs, at least 5 cm long and documented by
ultrasonographic investigation or other objective methods. Treatment should be initiated as soon as
possible following diagnosis and after exclusion of concomitant DVT or superficial-vein thrombosis
within 3 cm from the sapheno-femoral junction. Treatment should be continued for a minimum of 30
days and up to a maximum of 45 days in patients at high risk of thromboembolic complications (see
sections 4.4 and 5.1). Patients could be recommended to self-inject the product when they are judged
willing and able to do so. Physicians should provide clear instructions for self-injection.
•
P
atients who are to undergo surgery or other invasive procedures
In superficial vein thrombosis patients who are to undergo surgery or other invasive procedures,
fondaparinux, where possible, should not be given during the 24 hours before surgery.
Fondaparinux may be restarted at least 6 hours post-operatively provided haemostasis has been
achieved.
Special populations
In patients undergoing surgery, timing of the first fondaparinux injection requires strict adherence in
patients ≥75 years, and/or with body weight <50 kg and/or with renal impairment with creatinine
clearance ranging between 20 to 50 ml/min.
The first fondaparinux administration should be given not earlier than 6 hours following surgical
closure. The injection should not be given unless haemostasis has been established (see section 4.4).
Renal impairment
•
Prevention of VTE
- Fondaparinux should not be used in patients with creatinine clearance <20
ml/min (see section 4.3). The dose should be reduced to 1.5 mg once daily in patients with
creatinine clearance in the range of 20 to 50 ml/min (see sections 4.4 and 5.2). No dosage
reduction is required for patients with mild renal impairment (creatinine clearance >50
ml/min).
•
Treatment of superficial-vein thrombosis
- Fondaparinux should not be used in patients with
creatinine clearance <20 ml/min (see section 4.3). The dose should be reduced to 1.5 mg once
daily in patients with creatinine clearance in the range of 20 to 50 ml/min (see sections 4.4 and
5.2). No dosage reduction is required for patients with mild renal impairment (creatinine
clearance >50 ml/min). The safety and efficacy of 1.5 mg has not been studied (see section
4.4.)
Hepatic impairment
•
Prevention of VTE
- No dosing adjustment is necessary in patients with either mild or moderate
hepatic impairment. In patients with severe hepatic impairment, fondaparinux should be used
with care as this patient group has not been studied (see sections 4.4 and 5.2).
•
Treatment of superficial-vein thrombosis
- The safety and efficacy of fondaparinux in patients
with severe hepatic impairment has not been studied, therefore fondaparinux is not
recommended for use in these patients (see section 4.4).
Paediatric population -
Fondaparinux is not recommended for use in children below 17 years of age
due to a lack of data on safety and efficacy.
3
Low body weight
•
Prevention of VTE
- Patients with body weight <50 kg are at increased risk of bleeding.
Elimination of fondaparinux decreases with weight. Fondaparinux should be used with caution
in these patients (see section 4.4).
•
Treatment of superficial-vein thrombosis
- The safety and efficacy of fondaparinux in patients
with body weight less than 50 kg has not been studied
,
therefore fondaparinux is not
recommended for use in these patients (see section 4.4).
Method of administration
Fondaparinux is administered by deep subcutaneous injection while the patient is lying down. Sites of
administration should alternate between the left and the right anterolateral and left and right
posterolateral abdominal wall. To avoid the loss of medicinal product when using the pre-filled
syringe do not expel the air bubble from the syringe before the injection. The whole length of the
needle should be inserted perpendicularly into a skin fold held between the thumb and the forefinger;
the skin fold should be held throughout the injection.
For additional instructions for use and handling and disposal see section 6.6.
-
hypersensitivity to the active substance or to any of the excipients
-
active clinically significant bleeding
-
severe renal impairment defined by creatinine clearance < 20 ml/min.
Fondaparinux is intended for subcutaneous use only. Do not administer intramuscularly
.
Haemorrhage
Fondaparinux
should be used with caution in patients who have an increased risk of haemorrhage,
such as those with congenital or acquired bleeding disorders (e.g. platelet count <50,000/mm
3
), active
ulcerative gastrointestinal disease and recent intracranial haemorrhage or shortly after brain, spinal or
ophthalmic surgery and in special patient groups as outlined below.
•
For prevention of VTE
- Agents that may enhance the risk of haemorrhage should not be
administered concomitantly with fondaparinux. These agents include desirudin, fibrinolytic
agents, GP IIb/IIIa receptor antagonists, heparin, heparinoids, or Low Molecular Weight
Heparin (LMWH). When required, concomitant therapy with vitamin K antagonist should be
administered in accordance with the information of Section 4.5. Other antiplatelet medicinal
products (acetylsalicylic acid, dipyridamole, sulfinpyrazone, ticlopidine or clopidogrel), and
NSAIDs should be used with caution. If co-administration is essential, close monitoring is
necessary.
•
For treatment of superficial-vein thrombosis
- Fondaparinux should be used with caution in
patients who are being treated concomitantly with other medicinal products that increase the
risk of haemorrhage.
Patients with superficial-vein thrombosis
Presence of superficial-vein thrombosis greater than 3 cm from the sapheno-femoral junction should
be confirmed and concomitant DVT should be excluded by compression ultrasound or objective
methods prior to initiating treatment with fondaparinux. There are no data regarding the use of
fondaparinux 2.5 mg in superficial-vein thrombosis patients with concomitant DVT or with
superficial-vein thrombosis within 3 cm of the sapheno-femoral junction (see section 4.2 and 5.1).
4
-
acute bacterial endocarditis
The safety and efficacy of fondaparinux 2.5 mg has not been studied in the following groups: patients
with superficial-vein thrombosis following sclerotherapy or resulting as a complication of an
intravenous line, patients with history of superficial-vein thrombosis within the previous 3 months,
patients with history of venous thromboembolic disease within the previous 6 months,
or patients with active cancer (see section 4.2 and 5.1).
Spinal / Epidural anaesthesia
In patients undergoing major orthopaedic surgery, epidural or spinal haematomas that may result in
long-term or permanent paralysis cannot be excluded with the concurrent use of fondaparinux
and
spinal/epidural anaesthesia or spinal puncture. The risk of these rare events may be higher with post-
operative use of indwelling epidural catheters or the concomitant use of other medicinal products
affecting haemostasis.
Elderly patients
The elderly population is at increased risk of bleeding. As renal function is generally decreasing with
age, elderly patients may show reduced elimination and increased exposure of fondaparinux (see
section 5.2). Fondaparinux should be used
with caution in elderly patients (see section 4.2).
Low body weight
•
Prevention of VTE -
Patients with body weight <50 kg are at increased risk of bleeding.
Elimination of fondaparinux decreases with weight. Fondaparinux should be used with caution
in these patients (see section 4.2).
•
Treatment of superficial-vein thrombosis
- There are no clinical data available for the use of
fondaparinux for the treatment of superficial-vein thrombosis in patients with body weight less
than 50kg. Therefore, fondaparinux is not recommended for treatment of superficial-vein
thrombosis in these patients (see section 4.2).
Renal impairment
•
Prevention of VTE -
Fondaparinux is known to be mainly excreted by the kidney. Patients with
creatinine clearance <50 ml/min are at increased risk of bleeding and VTE and should be
treated with caution (see sections 4.2, 4.3 and 5.2). There are limited clinical data available
from patients with creatinine clearance less than 30 ml/min.
•
Treatment of superficial-vein thrombosis
- Fondaparinux should not be used in patients with
creatinine clearance <20 ml/min (see section 4.3). The dose should be reduced to 1.5 mg once
daily in patients with creatinine clearance in the range of 20 to 50 ml/min (see sections 4.2 and
5.2). The safety and efficacy of 1.5 mg has not been studied.
Severe hepatic impairment
•
Prevention of VTE -
Dosing adjustment of fondaparinux is not necessary. However, the use of
fondaparinux should be considered with caution because of an increased risk of bleeding due
to a deficiency of coagulation factors in patients with severe hepatic impairment (see section
4.2).
•
Treatment of superficial-vein thrombosis
- There are no clinical data available for the use of
fondaparinux for the treatment of superficial-vein thrombosis in patients with severe hepatic
impairment. Therefore, fondaparinux is not recommended for the treatment of superficial-vein
thrombosis in these patients (see section 4.2).
Patients with Heparin Induced Thrombocytopenia
Fondaparinux should be used with caution in patients with a history of HIT. The efficacy and safety
of fondaparinux have not been formally studied in patients with HIT type II. Fondaparinux does not
5
bind to platelet factor 4 and does not cross-react with sera from patients with Heparin Induced
Thrombocytopenia (HIT) type II. However, rare spontaneous reports of HIT in patients treated with
fondaparinux have been received. To date a causal association between treatment with fondaparinux
and the occurrence of HIT has not been established.
Bleeding risk is increased with concomitant administration of fondaparinux and agents that may
enhance the risk of haemorrhage (see section 4.4).
Oral anticoagulants (warfarin), platelet inhibitors (acetylsalicylic acid), NSAIDs (piroxicam) and
digoxin did not interact with the pharmacokinetics of fondaparinux. The fondaparinux dose (10 mg) in
the interaction studies was higher than the dose recommended for the present indications.
Fondaparinux neither influenced the INR activity of warfarin, nor the bleeding time under
acetylsalicylic acid or piroxicam treatment, nor the pharmacokinetics of digoxin at steady state.
Follow-up therapy with another anticoagulant medicinal product
If follow-up treatment is to be initiated with heparin or LMWH, the first injection should, as a general
rule, be given one day after the last fondaparinux injection.
If follow up treatment with a Vitamin K antagonist is required, treatment with fondaparinux should be
continued until the target INR value has been reached.
Pregnancy
There are no adequate data from the use of fondaparinux in pregnant women. Animal studies are
insufficient with respect to effects on pregnancy, embryo/foetal development, parturition and postnatal
development because of limited exposure. Fondaparinux should not be prescribed to pregnant women
unless clearly necessary.
Breastfeeding
Fondaparinux is excreted in rat milk but it is not known whether fondaparinux is excreted in human
milk. Breastfeeding is not recommended during treatment with fondaparinux. Oral absorption by the
child is however unlikely.
Fertility
There are no data available on the effect of fondaparinux on human fertility. Animal studies do not
show any effect on fertility.
No studies on the effect on the ability to drive and to use machines have been performed.
The most commonly reported serious adverse reactions reported with fondaparinux are bleeding
complications (various sites including rare cases of intracranial/ intracerebral and retroperitoneal
bleedings) and anaemia. Fondaparinux should be used with caution in patients who have an increased
risk of haemorrhage (see section 4.4).
The safety of
fondaparinux 2.5 mg
has been evaluated in 3,595 patients undergoing major orthopaedic
surgery of the lower limbs treated up to 9 days, in 327 patients undergoing hip fracture surgery treated
for 3 weeks following an initial prophylaxis of 1 week, 1,407 patients undergoing abdominal surgery
treated up to 9 days, and in 425 medical patients who are at risk for thromboembolic complications
treated up to 14 days.
6
The adverse reactions reported by the investigator as at least possibly related to fondaparinux are
presented within each frequency grouping (very common ≥ 1/10; common: ≥1/100 to < 1/10;
uncommon: ≥ 1/1,000 to < 1/100; rare: ≥ 1/10,000 to <1/1,000; very rare <1/10,000) and system organ
class by decreasing order of seriousness; these adverse reactions should be interpreted within the
surgical and medical context.
System organ class
MedDRA
Adverse reactions in patients
undergoing major orthopaedic
surgery of lower limbs and/or
abdominal surgery
Adverse reactions in medical
patients
Infections and
infestations
Rare:
post-operative wound
infection
Blood and lymphatic
system disorders
Common:
post-operative
haemorrhage, anaemia
Uncommon:
bleeding (epistaxis,
gastrointestinal, haemoptysis,
haematuria, haematoma)
thrombocytopenia, purpura,
thrombocythaemia, platelet
abnormal, coagulation disorder
Common:
bleeding
(haematoma, haematuria,
haemoptysis, gingival bleeding)
Uncommon:
anaemia
Immune system disorders Rare:
allergic reaction
Metabolism and nutrition
disorders
Rare:
hypokalaemia
Nervous system disorders Rare:
anxiety, somnolence,
vertigo, dizziness, headache,
confusion
Vascular disorders
Rare:
hypotension
Respiratory, thoracic and
mediastinal disorders
Rare:
dyspnoea, coughing
Uncommon:
dyspnoea
Gastrointestinal
disorders
Uncommon:
nausea, vomiting
Rare:
abdominal pain, dyspepsia,
gastritis, constipation, diarrhoea
Hepatobiliary disorders Uncommon:
hepatic enzymes
increased, hepatic function
abnormal
Rare:
bilirubinaemia
7













Skin and subcutaneous
tissue disorders
Uncommon:
rash, pruritus
Uncommon
: rash, pruritus
General disorders and
administration site
conditions
Uncommon:
oedema, oedema
peripheral, fever, wound
secretion
Rare:
chest pain, fatigue, hot
flushes, leg pain, oedema genital,
flushing, syncope
Uncommon
: chest pain
In other studies or in post-marketing experience, rare cases of intracranial / intracerebral and
retroperitoneal bleedings have been reported.
Fondaparinux doses above the recommended regimen
may lead to an increased risk of bleeding. There
is no known antidote to fondaparinux.
for the primary cause. Initiation of appropriate therapy such as surgical haemostasis, blood
replacements, fresh plasma transfusion, plasmapheresis should be considered.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antithrombotic agents.
ATC code: B01AX05
Pharmacodynamic effects
Fondaparinux is a synthetic and selective inhibitor of activated Factor X (Xa). The antithrombotic
activity of fondaparinux is the result of antithrombin III (ATIII) mediated selective inhibition of
Factor Xa. By binding selectively to ATIII, fondaparinux potentiates (about 300 times) the innate
neutralization of Factor Xa by ATIII. Neutralisation of Factor Xa interrupts the blood coagulation
cascade and inhibits both thrombin formation and thrombus development. Fondaparinux does not
inactivate thrombin (activated Factor II) and has no effects on platelets.
At the 2.5 mg dose, fondaparinux does not affect routine coagulation tests such as activated partial
thromboplastin time (aPTT), activated clotting time (ACT) or prothrombin time (PT)/International
Normalised Ratio (INR) tests in plasma nor bleeding time or fibrinolytic activity. However, rare
spontaneous reports of aPTT prolongation have been received.
Fondaparinux does not cross-react with sera from patients with heparin-induced thrombocytopaenia.
Clinical studies
Prevention of Venous Thromboembolic Events (VTE) in patients undergoing major orthopaedic
surgery of the lower limbs treated up to 9 days
The fondaparinux
clinical program was designed to demonstrate the efficacy of fondaparinux for the
prevention of venous thromboembolic events (VTE), i.e. proximal and distal deep vein thrombosis
(DVT) and pulmonary embolism (PE) in patients undergoing major orthopaedic surgery of the lower
limbs such as hip fracture, major knee surgery or hip replacement surgery. Over 8,000 patients (hip
fracture – 1,711, hip replacement – 5,829, major knee surgery – 1,367) were studied in controlled
8






Phase II and III clinical studies. Fondaparinux 2.5 mg once daily started 6-8 hours postoperatively was
compared with enoxaparin 40 mg once daily started 12 hours before surgery, or 30 mg twice daily
started 12-24 hours after surgery.
In a pooled analysis of these studies, the recommended dose regimen of fondaparinux versus
enoxaparin was associated with a significant decrease (54% [95% CI, 44 %; 63%]) in the rate of VTE
evaluated up to day 11 after surgery, irrespective of the type of surgery performed. The majority of
endpoint events were diagnosed by a prescheduled venography and consisted mainly of distal DVT,
but the incidence of proximal DVT was also significantly reduced. The incidence of symptomatic
VTE, including PE was not significantly different between treatment groups.
In studies versus enoxaparin 40 mg once daily started 12 hours before surgery, major bleeding was
observed in 2.8% of fondaparinux patients treated with the recommended dose, compared to 2.6%
with enoxaparin.
Prevention of Venous Thromboembolic Events (VTE) in patients undergoing hip fracture
surgery treated for up to 24 days following an initial prophylaxis of 1 week
In a randomised double-blind clinical trial, 737 patients were treated with fondaparinux 2.5 mg once
daily for 7 +/- 1 days following hip fracture surgery. At the end of this period, 656 patients were
randomised to receive fondaparinux 2.5 mg once daily or placebo for an additional 21 +/- 2 days.
Fondaparinux provided a significant reduction in the overall rate of VTE compared with placebo [3
patients (1.4%) vs 77 patients (35%), respectively]. The majority (70/80) of the recorded VTE events
were venographically detected non-symptomatic cases of DVT. Fondaparinux also provided a
significant reduction in the rate of symptomatic VTE (DVT, and / or PE) [1 (0.3%) vs 9 (2.7%)
patients, respectively] including two fatal PE reported in the placebo group. Major bleedings, all at
surgical site and none fatal, were observed in 8 patients (2.4%) treated with fondaparinux 2.5 mg
compared to 2 (0.6%) with placebo.
Prevention of Venous Thromboembolic Events (VTE) in patients undergoing abdominal surgery
who are judged to be at high risk of thromboembolic complications, such as patients undergoing
abdominal cancer surgery
In a double-blind clinical study, 2,927 patients were randomized to receive fondaparinux 2.5mg once
daily or dalteparin 5,000 IU once daily, with one 2,500 IU preoperative injection and a first 2,500 IU
post-operative injection, for 7+2 days. The main sites of surgery were colonic/rectal, gastric, hepatic,
cholecystectomy or other biliary. Sixty-nine percent of the patients underwent surgery for cancer.
Patients under-going urological (other than kidney) or gynaecological surgery, laparoscopic surgery or
vascular surgery were not included in the study.
In this study, the incidence of total VTE was 4.6% (47/1,027) with fondaparinux, versus 6.1%:
(62/1,021) with dalteparin: odds ratio reduction [95%CI] = -25.8% [-49.7%, 9.5%]. The difference in
total VTE rates between the treatment groups, which was not statistically significant, was mainly due
to a reduction of asymptomatic distal DVT. The incidence of symptomatic DVT was similar between
treatment groups: 6 patients (0.4%) in the fondaparinux group vs 5 patients (0.3%) in the dalteparin
group. In the large subgroup of
patients undergoing cancer surgery (69% of the patient population),
the VTE rate was 4.7% in the fondaparinux group, versus 7.7% in the dalteparin group.
Major bleeding was observed in 3.4% of the patients in the fondaparinux group and in 2.4% of the
dalteparin group.
Prevention of Venous Thromboembolic Events (VTE) in medical patients who are at high risk
for thromboembolic complications due to restricted mobility during acute illness
In a randomised double-blind clinical trial, 839 patients were treated with fondaparinux 2.5 mg once
daily or placebo for 6 to 14 days. This study included acutely ill medical patients, aged ≥ 60 years,
expected to require bed rest for at least four days, and hospitalized for congestive heart failure NYHA
class III/IV and/or acute respiratory illness and/or acute infectious or inflammatory disease.
Fondaparinux significantly reduced the overall rate of VTE compared to placebo [18 patients (5.6%)
vs 34 patients (10.5%), respectively]. The majority of events were asymptomatic distal DVT.
9
Fondaparinux also significantly reduced the rate of adjudicated fatal PE [0 patients (0.0%) vs 5
patients (1.2%), respectively]. Major bleedings were observed in 1 patient (0.2%) of each group.
Treatment of patients with acute symptomatic spontaneous superficial-vein thrombosis without
concomitant Deep-Vein Thrombosis (DVT)
A randomized, double blind, clinical trial (CALISTO) included 3002 patients with acute symptomatic
isolated, spontaneous superficial-vein thrombosis of the lower limbs, at least 5 cm long, confirmed by
compression ultrasonography.
Patients were not included if they had concomitant DVT or superficial-
vein thrombosis
within 3 cm of the sapheno-femoral junction. Patients were excluded if they had
severe hepatic impairment, severe renal impairment (creatinine clearance <30ml/min), low body
weight (<50kg), active cancer, symptomatic PE or a recent history of DVT/PE (<6 months) or
superficial-vein thrombosis (<90 days), or superficial-vein thrombosis associated with sclerotherapy or
a complication of an IV line, or they were at high risk of bleeding.
Patients were randomized to receive fondaparinux 2.5 mg once daily or placebo for 45 days in
addition to elastic stockings, analgesic and/or topical NSAIDS anti-inflammatory drugs. Follow-up
continued up to Day 77. The study population was 64% female, with a median age of 58 years, 4.4%
had a creatinine clearance <50ml/min.
The primary efficacy outcome, a composite of symptomatic PE, symptomatic DVT, symptomatic
superficial-vein thrombosis extension, symptomatic superficial-vein thrombosis reoccurrence, or
Death up to Day 47, was significantly reduced from 5.9% in placebo patients to 0.9% in those
receiving fondaparinux 2.5 mg (relative risk reduction: 85.2%; 95% CIs, 73.7% to 91.7% [p<0.001]).
The incidence of each thromboembolic component of the primary outcome was also significantly
reduced in fondaparinux patients as follows: symptomatic PE [0 (0%) vs 5 (0.3%) (p=0.031)],
symptomatic DVT [3 (0.2%) vs 18 (1.2%); relative risk reduction 83.4% (p<0.001)], symptomatic
superficial-vein thrombosis extension [4 (0.3%) vs 51 (3.4%); relative risk reduction 92.2%
(p<0.001)], symptomatic superficial-vein thrombosis reoccurrence [5 (0.3%) vs 24 (1.6%); relative
risk reduction 79.2% (p<0.001)].
The mortality rates were low and similar between the treatments groups with
2 (0.1%) deaths in
the fondaparinux group versus 1 (0.1%) death in the placebo group.
Efficacy was maintained up to Day 77 and was consistent across all predefined subgroups including
patients with varicose veins and patients with superficial-vein thrombosis located below the knee.
Major bleeding during treatment occurred in 1 (0.1%) fondaparinux patient and in 1 (0.1%) placebo
patient. Clinically relevant non major bleeding occurred in 5 (0.3%) fondaparinux patients and 8
(0.5%) placebo patients.
5.2 Pharmacokinetic properties
Absorption
After subcutaneous dosing, fondaparinux is completely and rapidly absorbed (absolute bioavailability
100%). Following a single subcutaneous injection of fondaparinux
2.5 mg to young healthy subjects,
peak plasma concentration (mean C
max
= 0.34 mg/l) is obtained 2 hours post-dosing. Plasma
concentrations of half the mean C
max
values are reached 25 minutes post-dosing.
In elderly healthy subjects, pharmacokinetics of fondaparinux are linear in the range of 2 to 8 mg by
subcutaneous route. Following once daily dosing, steady state of plasma levels is obtained after 3 to 4
days with a 1.3-fold increase in C
max
and AUC.
Mean (CV%) steady state pharmacokinetic parameters estimates of fondaparinux in patients
undergoing hip replacement surgery receiving fondaparinux
2.5 mg once daily are: C
max
(mg/l) - 0.39
(31%), T
max
(h) - 2.8 (18%) and C
min
(mg/l) -0.14 (56%). In hip fracture patients, associated with their
10
increased age, fondaparinux steady state plasma concentrations are: C
max
(mg/l) - 0.50 (32%),
C
min
(mg/l) - 0.19 (58%).
Distribution
The distribution volume of fondaparinux is limited (7-11 litres).
In vitro
, fondaparinux is highly and
specifically bound to antithrombin protein with a dose-dependant plasma concentration binding
(98.6% to 97.0% in the concentration range from 0.5 to 2 mg/l). Fondaparinux does not bind
significantly to other plasma proteins, including platelet factor 4 (PF4).
Since fondaparinux does not bind significantly to plasma proteins other than ATIII, no interaction
with other medicinal products by protein binding displacement are expected.
Biotransformation
Although not fully evaluated, there is no evidence of fondaparinux metabolism and in particular no
evidence for the formation of active metabolites.
Fondaparinux does not inhibit CYP450s (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6,
CYP2E1 or CYP3A4)
in vitro
. Thus, fondaparinux is not expected to interact with other medicinal
products
in vivo
by inhibition of CYP-mediated metabolism.
Excretion/Elimination
The elimination half-life (t
½
) is about 17 hours in healthy young subjects and about 21 hours in healthy
elderly subjects. Fondaparinux is excreted to 64 – 77 % by the kidney as unchanged compound.
Special populations
Paediatric patients
- Fondaparinux
has not been investigated in this population for the prevention of
VTE or for the treatment of superficial vein thrombosis.
Elderly patients
- Renal function may decrease with age and thus, the elimination capacity for
fondaparinux may be reduced in elderly.
In patients >75 years undergoing orthopaedic surgery, the
estimated plasma clearance was 1.2 to 1.4 times lower than in patients <65 years.
Renal impairment
- Compared with patients with normal renal function (creatinine
clearance > 80 ml/min), plasma clearance is 1.2 to 1.4 times lower in patients with mild renal
impairment (creatinine clearance 50 to 80 ml/min) and on average 2 times lower in patients with
moderate renal impairment (creatinine clearance 30 to 50 ml/min). In severe renal impairment
(creatinine clearance < 30 ml/min), plasma clearance is approximately 5 times lower than in normal
renal function. Associated terminal half-life values were 29 h in moderate and 72 h in patients with
severe renal impairment.
Gender
- No gender differences were observed after adjustment for body weight.
Race
- Pharmacokinetic differences due to race have not been studied prospectively. However, studies
performed in Asian (Japanese) healthy subjects did not reveal a different pharmacokinetic profile
compared to Caucasian healthy subjects. Similarly, no plasma clearance differences were observed
between black and Caucasian patients undergoing orthopaedic surgery.
Body weight
-
Plasma clearance of fondaparinux increases with body weight (9% increase per 10 kg).
Hepatic impairment -
Following a single, subcutaneous dose of fondaparinux in subjects with
moderate hepatic impairment (Child-Pugh Category B), total (i.e., bound and unbound) C
max
and AUC
were decreased by 22% and 39%, respectively, as compared to subjects with normal liver function.
The lower plasma concentrations of fondaparinux were attributed to reduced binding to ATIII
secondary to the lower ATIII plasma concentrations in subjects with hepatic impairment thereby
resulting in increased renal clearance of fondaparinux. Consequently, unbound concentrations of
11
fondaparinux are expected to be unchanged in patients with mild to moderate hepatic impairment, and
therefore, no dose adjustment is necessary based on pharmacokinetics.
The pharmacokinetics of fondaparinux has not been studied in patients with severe hepatic impairment
(see sections 4.2 and 4.4).
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity, and genotoxicity. Animal studies are insufficient with respect to
effects on toxicity to reproduction because of limited exposure.
6.
6.1 List of excipients
Sodium chloride
Water for injections
Hydrochloric acid
Sodium hydroxide
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3 Shelf life
3 years.
6.4 Special precautions for storage
Do not freeze.
6.5 Nature and contents of container
Type I glass barrel (1 ml) affixed with a 27 gauge x 12.7 mm needle and stoppered with a bromobutyl
or chlorobutyl elastomer plunger stopper.
Arixtra
is available in pack sizes of 2, 7, 10 and 20 pre-filled syringes. There are two types of syringes:
•
syringe with a yellow plunger and an automatic safety system
•
syringe with yellow plunger and a manual safety system.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
The subcutaneous injection is administered in the same way as with a classical syringe.
Parenteral solutions should be inspected visually for particulate matter and discoloration prior to
administration.
Instruction for self-administration is mentioned in the Package Leaflet.
The needle protection system of the Arixtra pre-filled syringes have been designed with a safety
system to protect from needle stick injuries following injection.
12
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Glaxo Group Ltd
Greenford
Middlesex
UB6 0NN
United Kingdom
8.
EU/1/02/206/005-008
EU/1/02/206/024
EU/1/02/206/025
EU/1/02/206/026
9.
Date of first authorisation: 21 March 2002
Date of latest renewal: 21 March 2007
Detailed information on this medicinal product is available on the website of the European Medicines
Agency http://www.ema.europa.eu
13
1.
Arixtra 2.5 mg/0.5 ml solution for injection, pre-filled syringe.
2.
Each pre-filled syringe (0.5 ml) contains 2.5 mg of fondaparinux sodium.
Excipient(s): Contains less than 1 mmol of sodium (23 mg) per dose, and therefore is essentially
sodium free.
For a full list of excipients, see section 6.1.
3.
Solution for injection.
The solution is a clear and colourless liquid.
4.
Prevention of Venous Thromboembolic Events (VTE) in adults undergoing major orthopaedic surgery
of the lower limbs such as hip fracture, major knee surgery or hip replacement surgery.
Prevention of Venous Thromboembolic Events (VTE) in adults undergoing abdominal surgery who
are judged to be at high risk of thromboembolic complications, such as patients undergoing abdominal
cancer surgery (see section 5.1).
Prevention of Venous Thromboembolic Events (VTE) in adult medical patients who are judged to be
at high risk for VTE and who are immobilised due to acute illness such as cardiac insufficiency and/or
acute respiratory disorders, and/or acute infectious or inflammatory disease.
Treatment of unstable angina or non-ST segment elevation myocardial infarction (UA/NSTEMI) in
adultsfor whom urgent (< 120 mins) invasive management (PCI) is not indicated (see sections 4.4
and 5.1).
Treatment of ST segment elevation myocardial infarction (STEMI) in adults who are managed with
thrombolytics or who initially are to receive no other form of reperfusion therapy.
Treatment of adults with acute symptomatic spontaneous superficial-vein thrombosis of the lower
limbs without concomitant deep-vein thrombosis (see sections 4.2 and 5.1).
Posology
Patients undergoing major orthopaedic or abdominal surgery
The recommended dose of fondaparinux
is 2.5 mg once daily administered post-operatively by
subcutaneous injection.
The initial dose should be given
6 hours following surgical closure provided that haemostasis has been
established.
14
Treatment should be continued until the risk of venous thrombo-embolism has diminished, usually
until the patient is ambulant, at least 5 to 9 days after surgery. Experience shows that in patients
undergoing hip fracture surgery, the risk of VTE continues beyond 9 days after surgery. In these
patients the use of prolonged prophylaxis with fondaparinux should be considered for up to an
additional 24 days (see section 5.1).
Medical patients who are at high risk for thromboembolic complications based on an individual risk
assessment
The recommended dose of fondaparinux is 2.5 mg once daily administered by subcutaneous injection.
A treatment duration of 6-14 days has been clinically studied in medical patients (see section 5.1).
Treatment of unstable angina/non- ST segment elevation myocardial infarction (UA/NSTEMI)
The recommended dose of fondaparinux is 2.5 mg once daily, administered by subcutaneous injection.
Treatment should be initiated as soon as possible following diagnosis and continued for up to a
maximum of 8 days or until hospital discharge if that occurs earlier.
If a patient is to undergo percutaneous coronary intervention (PCI), unfractionated heparin (UFH) as
per local practice should be administered during PCI, taking into account the patient’s potential risk of
bleeding, including the time since the last dose of fondaparinux (see section 4.4). The timing of
restarting subcutaneous fondaparinux after sheath removal should be based on clinical judgment. In
the pivotal UA/NSTEMI clinical trial, treatment with fondaparinux was restarted no earlier than 2
hours after sheath removal.
Treatment of ST segment elevation myocardial infarction (STEMI)
The recommended dose of fondaparinux is 2.5 mg once daily. The first dose of fondaparinux is
administered intravenously and subsequent doses are administered by subcutaneous injection.
Treatment should be initiated as soon as possible following diagnosis and continued for up to a
maximum of 8 days or until hospital discharge if that occurs earlier.
If a patient is to undergo non-primary PCI, unfractionated heparin (UFH) as per local practice should
be administered during PCI, taking into account the patient’s potential risk of bleeding, including the
time since the last dose of fondaparinux (see section 4.4). The timing of restarting subcutaneous
fondaparinux after sheath removal should be based on clinical judgment. In the pivotal STEMI clinical
trial, treatment with fondaparinux was restarted no earlier than 3 hours after sheath removal.
•
Patients who are to undergo coronary artery bypass graft (CABG) surgery
In STEMI or UA/NSTEMI patients who are to undergo coronary artery bypass graft (CABG)
surgery, fondaparinux where possible, should not be given during the 24 hours before surgery and
may be restarted 48 hours post-operatively
.
Treatment of superficial-vein thrombosis
The recommended dose of fondaparinux is 2.5 mg once daily, administered by subcutaneous injection.
Patients eligible for fondaparinux 2.5 mg treatment should have acute, symptomatic, isolated,
spontaneous superficial-vein thrombosis of the lower limbs, at least 5 cm long and documented by
ultrasonographic investigation or other objective methods. Treatment should be initiated as soon as
possible following diagnosis and after exclusion of concomitant DVT or superficial-vein thrombosis
within 3 cm from the sapheno-femoral junction. Treatment should be continued for a minimum of 30
days and up to a maximum of 45 days in patients at high risk of thromboembolic complications (see
sections 4.4 and 5.1).
Patients could be recommended to self-inject the product when they are judged
willing and able to do so. Physicians should provide clear instructions for self-injection.
•
P
atients who are to undergo surgery or other invasive procedures
In superficial vein thrombosis patients who are to undergo surgery or other invasive procedures,
fondaparinux, where possible, should not be given during the 24 hours before surgery.
Fondaparinux may be restarted at least 6 hours post-operatively provided haemostasis has been
achieved.
15
Special populations
Prevention of VTE following Surgery
In patients undergoing surgery, timing of the first fondaparinux injection requires strict adherence in
patients ≥75 years, and/or with body weight <50 kg and/or with renal impairment with creatinine
clearance ranging between 20 to 50 ml/min.
The first fondaparinux administration should be given not earlier than 6 hours following surgical
closure. The injection should not be given unless haemostasis has been established (see section 4.4).
Renal impairment
•
Prophylaxis of VTE -
Fondaparinux should not be used in patients with creatinine
clearance <20 ml/min (see section 4.3). The dose should be reduced to 1.5 mg once daily in
patients with creatinine clearance in the range of 20 to 50 ml/min (see sections 4.4 and 5.2).
No dosage reduction is required for patients with mild renal impairment (creatinine clearance
>50 ml/min).
•
Treatment of UA/NSTEMI and
STEMI - Fondaparinux should not be used in patients with
creatinine clearance < 20 ml/min (see section 4.3). No dosage reduction is required for
patients with creatinine clearance > 20 ml/min.
•
Treatment of superficial-vein thrombosis
- Fondaparinux should not be used in patients with
creatinine clearance <20 ml/min (see section 4.3). The dose should be reduced to 1.5 mg once
daily in patients with creatinine clearance in the range of 20 to 50 ml/min (see sections 4.4 and
5.2). No dosage reduction is required for patients with mild renal impairment (creatinine
clearance >50 ml/min). The safety and efficacy of 1.5 mg has not been studied (see section
4.4.)
Hepatic impairment
•
Prevention of VTE and Treatment of UA/NSTEMI and STEMI
- No dosing adjustment is
necessary in patients with either mild or moderate hepatic impairment. In patients with severe
hepatic impairment, fondaparinux should be used with care as this patient group has not been
studied (see sections 4.4 and 5.2).
•
Treatment of superficial-vein thrombosis
- The safety and efficacy of fondaparinux in patients
with severe hepatic impairment has not been studied, therefore fondaparinux is not
recommended for use in these patients (see section 4.4).
Paediatric population -
Fondaparinux is not recommended for use in children below 17 years of age
due to a lack of data on safety and efficacy.
Low body weight
•
Prevention of VTE and Treatment of UA/NSTEMI and STEMI
- Patients with body weight <50
kg are at increased risk of bleeding. Elimination of fondaparinux decreases with weight.
Fondaparinux should be used with caution in these patients (see section 4.4).
•
Treatment of superficial-vein thrombosis
- The safety and efficacy of fondaparinux in patients
with body weight less than 50 kg has not been studied
,
therefore fondaparinux is not
recommended for use in these patients (see section 4.4).
Method of administration
•
Subcutaneous administration
Fondaparinux is administered by deep subcutaneous injection while the patient is lying down.
Sites of administration should alternate between the left and the right anterolateral and left and
16
right posterolateral abdominal wall. To avoid the loss of medicinal product when using the pre-
filled syringe do not expel the air bubble from the syringe before the injection. The whole length
of the needle should be inserted perpendicularly into a skin fold held between the thumb and the
forefinger; the skin fold should be held throughout the injection.
•
Intravenous administration (first dose in patients with STEMI only)
Intravenous administration should be through an existing intravenous line either directly or using a
small volume (25 or 50ml) 0.9% saline minibag
.
To avoid the loss of medicinal product when
using the pre-filled syringe do not expel the air bubble from the syringe before the injection. The
intravenous tubing should be well flushed with saline after injection to ensure that all of the
medicinal product is administered. If administered via a minibag, the infusion should be given
over 1 to 2 minutes.
For additional instructions for use and handling and disposal see section 6.6.
-
hypersensitivity to the active substance or to any of the excipients
-
acute bacterial endocarditis
-
severe renal impairment defined by creatinine clearance < 20 ml/min.
Fondaparinux must not be administered intramuscularly
.
Haemorrhage
Fondaparinux
should be used with caution in patients who have an increased risk of haemorrhage,
such as those with congenital or acquired bleeding disorders (e.g. platelet count <50,000/mm
3
), active
ulcerative gastrointestinal disease and recent intracranial haemorrhage or shortly after brain, spinal or
ophthalmic surgery and in special patient groups as outlined below.
For prevention of VTE- Agents that may enhance the risk of haemorrhage should not be administered
concomitantly with fondaparinux. These agents include desirudin, fibrinolytic agents, GP IIb/IIIa
receptor antagonists, heparin, heparinoids, or Low Molecular Weight Heparin (LMWH). When
required, concomitant therapy with vitamin K antagonist should be administered in accordance with
the information of section 4.5. Other antiplatelet medicinal products (acetylsalicylic acid,
dipyridamole, sulfinpyrazone, ticlopidine or clopidogrel), and NSAIDs should be used with caution. If
co-administration is essential, close monitoring is necessary.
For treatment of UA/NSTEMI and STEMI
-Fondaparinux should be used with caution in patients who
are being treated concomitantly with other agents that increase the risk of haemorrhage (such as
GPIIb/IIIa inhibitors or thrombolytics).
For treatment of superficial-vein thrombosis -
Fondaparinux should be used with caution in patients
who are being treated concomitantly with other medicinal products that increase the risk of
haemorrhage
.
PCI and risk of guiding catheter thrombus
In STEMI patients undergoing primary PCI, the use of fondaparinux prior to and during PCI is not
recommended. Similarly, in UA/NSTEMI patients with life threatening conditions that require urgent
revascularisation, the use of fondaparinux prior to and during PCI is not recommended. These are
patients with refractory or recurrent angina associated with dynamic ST deviation, heart failure, life-
threatening arrhythmias or haemodynamic instability.
17
-
active clinically significant bleeding
In UA/NSTEMI and STEMI patients undergoing non-primary PCI, the use of fondaparinux as the sole
anticoagulant during PCI is not recommended, therefore UFH should be used according to local
practice (see section 4.2).
There are limited data on the use of UFH during non-primary PCI in patients treated with
fondaparinux (see section 5.1). In those patients who underwent non-primary PCI 6-24 hours after the
last dose of fondaparinux, the median dose of UFH was 8,000 IU and the incidence of major bleeding
was 2% (2/98). In those patients who underwent non-primary PCI <6 hours after the last dose of
fondaparinux, the median dose of UFH was 5,000 IU and the incidence of major bleeding was 4.1%
(2/49).
Clinical trials have shown a low but increased risk of guiding catheter thrombus in patients treated
with fondaparinux for anticoagulation during PCI compared to control. Incidences in non-primary PCI
in UA/NSTEMI were 1.0% vs 0.3% (fondaparinux vs. enoxaparin) and in primary PCI in STEMI were
1.2% vs 0% (fondaparinux vs. control).
Patients with superficial-vein thrombosis
Presence of superficial-vein thrombosis greater than 3 cm from the sapheno-femoral junction should
be confirmed and concomitant DVT should be excluded by compression ultrasound or objective
methods prior to initiating treatment of fondaparinux. There are no data regarding the use of
fondaparinux 2.5 mg in superficial-vein thrombosis patients with concomitant DVT or with
superficial-vein thrombosis within 3 cm of the sapheno-femoral junction (see section 4.2 and 5.1).
The safety and efficacy of fondaparinux 2.5 mg has not been studied in the following groups: patients
with superficial-vein thrombosis following sclerotherapy or resulting as a complication of an
intravenous line, patients with history of superficial-vein thrombosis within the previous 3 months,
patients with history of venous thromboembolic disease within the previous 6 months,
or patients with active cancer (see section 4.2 and 5.1).
Spinal / Epidural anaesthesia
In patients undergoing major orthopaedic surgery, epidural or spinal haematomas that may result in
long-term or permanent paralysis cannot be excluded with the concurrent use of fondaparinux
and
spinal/epidural anaesthesia or spinal puncture. The risk of these rare events may be higher with post-
operative use of indwelling epidural catheters or the concomitant use of other medicinal products
affecting haemostasis.
Elderly patients
The elderly population is at increased risk of bleeding. As renal function is generally decreasing with
age, elderly patients may show reduced elimination and increased exposure of fondaparinux (see
section 5.2). Fondaparinux should be used
with caution in elderly patients (see section 4.2).
Low body weight
•
Prevention of VTE and Treatment of UA/NSTEMI and STEMI
-
Patients with body weight <50 kg
are at increased risk of bleeding.
Elimination of fondaparinux decreases with weight.
Fondaparinux should be used with caution in these patients (see section 4.2).
•
Treatment of superficial-vein thrombosis
- There are no clinical data available for the use of
fondaparinux for the treatment of superficial-vein thrombosis in patients with body weight less
than 50kg. Therefore, fondaparinux is not recommended for treatment of superficial-vein
thrombosis in these patients (see section 4.2).
Renal impairment
Fondaparinux is known to be mainly excreted by the kidney.
•
Prophylaxis of VTE
- Patients with creatinine clearance <50 ml/min are at increased risk of
bleeding and VTE and should be treated with caution (see sections 4.2, 4.3 and 5.2). There are
limited clinical data available from patients with creatinine clearance less than 30 ml/min.
18
•
Treatment of UA/NSTEMI and
STEMI - For the treatment of UA/NSTEMI and STEMI, there are
limited clinical data available on the use of fondaparinux 2.5mg once daily in patients with
creatinine clearance between 20 and 30 ml/min. Therefore the physician should determine if the
benefit of treatment outweighs the risk (see sections 4.2 and 4.3).
•
Treatment of superficial-vein thrombosis
- Fondaparinux should not be used in patients with
creatinine clearance <20 ml/min (see section 4.3). The dose should be reduced to 1.5 mg once
daily in patients with creatinine clearance in the range of 20 to 50 ml/min (see sections 4.2 and
5.2). The safety and efficacy of 1.5 mg has not been studied.
Severe hepatic impairment
•
Prevention of VTE and Treatment of UA/NSTEMI and STEMI
-
Dosing adjustment of
fondaparinux is not necessary. However, the use of fondaparinux should be considered with
caution because of an increased risk of bleeding due to a deficiency of coagulation factors in
patients with severe hepatic impairment (see section 4.2).
•
Treatment of superficial-vein thrombosis
- There are no clinical data available for the use of
fondaparinux for the treatment of superficial-vein thrombosis in patients with severe hepatic
impairment. Therefore, fondaparinux is not recommended for the treatment of superficial-vein
thrombosis in these patients (see section 4.2).
Patients with Heparin Induced Thrombocytopenia
Fondaparinux should be used with caution in patients with a history of HIT. The efficacy and safety of
fondaparinux have not been formally studied in patients with HIT type II. Fondaparinux does not bind
to platelet factor 4 and does not cross-react with sera from patients with Heparin Induced
Thrombocytopenia (HIT) type II. However, rare spontaneous reports of HIT in patients treated with
fondaparinux have been received. To date a causal association between treatment with fondaparinux
and the occurrence of HIT has not been established.
Bleeding risk is increased with concomitant administration of fondaparinux and agents that may
enhance the risk of haemorrhage (see section 4.4).
Oral anticoagulants (warfarin), platelet inhibitors (acetylsalicylic acid), NSAIDs (piroxicam) and
digoxin did not interact with the pharmacokinetics of fondaparinux. The fondaparinux dose (10 mg) in
the interaction studies was higher than the dose recommended for the present indications.
Fondaparinux neither influenced the INR activity of warfarin, nor the bleeding time under
acetylsalicylic acid or piroxicam treatment, nor the pharmacokinetics of digoxin at steady state.
Follow-up therapy with another anticoagulant medicinal product
If follow-up treatment is to be initiated with heparin or LMWH, the first injection should, as a general
rule, be given one day after the last fondaparinux injection.
If follow up treatment with a Vitamin K antagonist is required, treatment with fondaparinux should be
continued until the target INR value has been reached.
Pregnancy
There are no adequate data from the use of fondaparinux in pregnant women. Animal studies are
insufficient with respect to effects on pregnancy, embryo/foetal development, parturition and postnatal
development because of limited exposure. Fondaparinux should not be prescribed to pregnant women
unless clearly necessary.
19
Breastfeeding
Fondaparinux is excreted in rat milk but it is not known whether fondaparinux is excreted in human
milk. Breastfeeding is not recommended during treatment with fondaparinux. Oral absorption by the
child is however unlikely.
Fertility
There are no data available on the effect of fondaparinux on human fertility. Animal studies do not
show any effect on fertility.
No studies on the effect on the ability to drive and to use machines have been performed.
The most commonly reported serious adverse reactions reported with fondaparinux are bleeding
complications (various sites including rare cases of intracranial/ intracerebral and retroperitoneal
bleedings) and anaemia. Fondaparinux should be used with caution in patients who have an increased
risk of haemorrhage (see section 4.4).
The safety of
fondaparinux 2.5 mg
has been evaluated in:
-
3,595 patients undergoing major orthopaedic surgery of the lower limbs treated up to 9 days
-
327 patients undergoing hip fracture surgery treated for 3 weeks following an initial prophylaxis
of 1 week
-
1,407 patients undergoing abdominal surgery treated up to 9 days
-
425 medical patients who are at risk for thromboembolic complications treated up to 14 days
-
10,057 patients undergoing treatment of UA or NSTEMI ACS
-
6,036 patients undergoing treatment of STEMI ACS.
For the prevention of VTE, the adverse reactions reported by the investigator as at least possibly
related to fondaparinux are presented within each frequency grouping (very common ≥ 1/10; common:
≥1/100 to < 1/10; uncommon: ≥ 1/1,000 to < 1/100; rare: ≥ 1/10,000 to <1/1,000; very rare <1/10,000)
and system organ class by decreasing order of seriousness; these adverse reactions should be
interpreted within the surgical and medical context.
System organ class
MedDRA
Adverse reactions in patients
undergoing major orthopaedic
surgery of lower limbs and/or
abdominal surgery
Adverse reactions in medical
patients
Infections and
infestations
Rare:
post-operative wound
infection
Blood and lymphatic
system disorders
Common:
post-operative
haemorrhage, anaemia
Uncommon:
bleeding (epistaxis,
gastrointestinal, haemoptysis,
haematuria, haematoma)
thrombocytopenia, purpura,
thrombocythaemia, platelet
abnormal, coagulation disorder
Common:
bleeding
(haematoma, haematuria,
haemoptysis, gingival bleeding)
Uncommon:
anaemia
20







Immune system disorders Rare:
allergic reaction
Metabolism and nutrition
disorders
Rare:
hypokalaemia
Nervous system disorders Rare:
anxiety, somnolence,
vertigo, dizziness, headache,
confusion
Vascular disorders
Rare:
hypotension
Respiratory, thoracic and
mediastinal disorders
Rare:
dyspnoea, coughing
Uncommon:
dyspnoea
Gastrointestinal
disorders
Uncommon:
nausea, vomiting
Rare:
abdominal pain, dyspepsia,
gastritis, constipation, diarrhoea
Hepatobiliary disorders Uncommon:
hepatic enzymes
increased, hepatic function
abnormal
Rare:
bilirubinaemia
Skin and subcutaneous
tissue disorders
Uncommon:
rash, pruritus
Uncommon
: rash, pruritus
General disorders and
administration site
conditions
Uncommon:
oedema, oedema
peripheral, fever, wound
secretion
Rare:
chest pain, fatigue, hot
flushes, leg pain, oedema genital,
flushing, syncope
Uncommon
: chest pain
In other studies or in post-marketing experience, rare cases of intracranial / intracerebral and
retroperitoneal bleedings have been reported.
The adverse event profile reported in the ACS program is consistent with the adverse drug reactions
identified for VTE prophylaxis.
Bleeding was a commonly reported event in patients with UA/NSTEMI and STEMI. The incidence of
adjudicated major bleeding was 2.1% (fondaparinux) vs. 4.1% (enoxaparin) up to and including Day 9
in the Phase III UA/NSTEMI study, and the incidence of adjudicated severe haemorrhage by modified
TIMI criteria was 1.1% (fondaparinux) vs. 1.4% (control [UFH/placebo]) up to and including Day 9 in
the Phase III STEMI study.
In the Phase III UA/NSTEMI study, the most commonly reported non-bleeding adverse events
(reported in at least 1% of subjects on fondaparinux) were headache, chest pain and atrial fibrillation.
In the Phase III study in STEMI patients, the most commonly reported non-bleeding adverse events
(reported in at least 1% of subjects on fondaparinux) were atrial fibrillation, pyrexia, chest pain,
headache, ventricular tachycardia, vomiting, and hypotension.
21













Fondaparinux doses above the recommended regimen
may lead to an increased risk of bleeding. There
is no known antidote to fondaparinux.
for the primary cause. Initiation of appropriate therapy such as surgical haemostasis, blood
replacements, fresh plasma transfusion, plasmapheresis should be considered.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antithrombotic agents.
ATC code: B01AX05
Pharmacodynamic effects
Fondaparinux is a synthetic and selective inhibitor of activated Factor X (Xa). The antithrombotic
activity of fondaparinux is the result of antithrombin III (ATIII) mediated selective inhibition of
Factor Xa. By binding selectively to ATIII, fondaparinux potentiates (about 300 times) the innate
neutralization of Factor Xa by ATIII. Neutralisation of Factor Xa interrupts the blood coagulation
cascade and inhibits both thrombin formation and thrombus development. Fondaparinux does not
inactivate thrombin (activated Factor II) and has no effects on platelets.
At the 2.5 mg dose, fondaparinux does not affect routine coagulation tests such as activated partial
thromboplastin time (aPTT), activated clotting time (ACT) or prothrombin time (PT)/International
Normalised Ratio (INR) tests in plasma nor bleeding time or fibrinolytic activity. However, rare
spontaneous reports of aPTT prolongation have been received.
Fondaparinux does not cross-react with sera from patients with heparin-induced thrombocytopaenia.
Clinical studies
Prevention of Venous Thromboembolic Events (VTE) in patients undergoing major orthopaedic
surgery of the lower limbs treated up to 9 days
The fondaparinux
clinical program was designed to demonstrate the efficacy of fondaparinux for the
prevention of venous thromboembolic events (VTE), i.e. proximal and distal deep vein thrombosis
(DVT) and pulmonary embolism (PE) in patients undergoing major orthopaedic surgery of the lower
limbs such as hip fracture, major knee surgery or hip replacement surgery. Over 8,000 patients (hip
fracture – 1,711, hip replacement – 5,829, major knee surgery – 1,367) were studied in controlled
Phase II and III clinical studies. Fondaparinux 2.5 mg once daily started 6-8 hours postoperatively was
compared with enoxaparin 40 mg once daily started 12 hours before surgery, or 30 mg twice daily
started 12-24 hours after surgery.
In a pooled analysis of these studies, the recommended dose regimen of fondaparinux versus
enoxaparin was associated with a significant decrease (54% [95% CI, 44 %; 63%]) in the rate of VTE
evaluated up to day 11 after surgery, irrespective of the type of surgery performed. The majority of
endpoint events were diagnosed by a prescheduled venography and consisted mainly of distal DVT,
but the incidence of proximal DVT was also significantly reduced. The incidence of symptomatic
VTE, including PE was not significantly different between treatment groups.
In studies versus enoxaparin 40 mg once daily started 12 hours before surgery, major bleeding was
observed in 2.8% of fondaparinux patients treated with the recommended dose, compared to 2.6%
with enoxaparin.
22
Prevention of Venous Thromboembolic Events (VTE) in patients undergoing hip fracture
surgery treated for up to 24 days following an initial prophylaxis of 1 week
In a randomised double-blind clinical trial, 737 patients were treated with fondaparinux 2.5 mg once
daily for 7 +/- 1 days following hip fracture surgery. At the end of this period, 656 patients were
randomised to receive fondaparinux 2.5 mg once daily or placebo for an additional 21 +/- 2 days.
Fondaparinux provided a significant reduction in the overall rate of VTE compared with placebo [3
patients (1.4%) vs 77 patients (35%), respectively]. The majority (70/80) of the recorded VTE events
were venographically detected non-symptomatic cases of DVT. Fondaparinux also provided a
significant reduction in the rate of symptomatic VTE (DVT, and / or PE) [1 (0.3%) vs 9 (2.7%)
patients, respectively] including two fatal PE reported in the placebo group. Major bleedings, all at
surgical site and none fatal, were observed in 8 patients (2.4%) treated with fondaparinux 2.5 mg
compared to 2 (0.6%) with placebo.
Prevention of Venous Thromboembolic Events (VTE) in patients undergoing abdominal surgery
who are judged to be at high risk of thromboembolic complications, such as patients undergoing
abdominal cancer surgery
In a double-blind clinical study, 2,927 patients were randomized to receive fondaparinux 2.5mg once
daily or dalteparin 5,000 IU once daily, with one 2,500 IU preoperative injection and a first 2,500 IU
post-operative injection, for 7+2 days. The main sites of surgery were colonic/rectal, gastric, hepatic,
cholecystectomy or other biliary. Sixty-nine percent of the patients underwent surgery for cancer.
Patients under-going urological (other than kidney) or gynaecological surgery, laparoscopic surgery or
vascular surgery were not included in the study.
In this study, the incidence of total VTE was 4.6% (47/1,027) with fondaparinux, versus 6.1%:
(62/1,021) with dalteparin: odds ratio reduction [95%CI] = -25.8% [-49.7%, 9.5%]. The difference in
total VTE rates between the treatment groups, which was not statistically significant, was mainly due
to a reduction of asymptomatic distal DVT. The incidence of symptomatic DVT was similar between
treatment groups: 6 patients (0.4%) in the fondaparinux group vs 5 patients (0.3%) in the dalteparin
group. In the large subgroup of
patients undergoing cancer surgery (69% of the patient population),
the VTE rate was 4.7% in the fondaparinux group, versus 7.7% in the dalteparin group.
Major bleeding was observed in 3.4% of the patients in the fondaparinux group and in 2.4% of the
dalteparin group.
Prevention of Venous Thromboembolic Events (VTE) in medical patients who are at high risk
for thromboembolic complications due to restricted mobility during acute illness
In a randomised double-blind clinical trial, 839 patients were treated with fondaparinux 2.5 mg once
daily or placebo for 6 to 14 days. This study included acutely ill medical patients, aged ≥ 60 years,
expected to require bed rest for at least four days, and hospitalized for congestive heart failure NYHA
class III/IV and/or acute respiratory illness and/or acute infectious or inflammatory disease.
Fondaparinux significantly reduced the overall rate of VTE compared to placebo [18 patients (5.6%)
vs 34 patients (10.5%), respectively]. The majority of events were asymptomatic distal DVT.
Fondaparinux also significantly reduced the rate of adjudicated fatal PE [0 patients (0.0%) vs 5
patients (1.2%), respectively]. Major bleedings were observed in 1 patient (0.2%) of each group.
Treatment of unstable angina or non-ST segment elevation myocardial infarction (UA/NSTEMI)
OASIS 5 was a double-blind, randomised, non-inferiority study with fondaparinux 2.5 mg
subcutaneously once daily versus enoxaparin 1 mg/kg subcutaneously twice daily in approximately
20,000 patients with UA/NSTEMI. All patients received standard medical treatment for UA/NSTEMI,
with 34% of patients undergoing PCI and 9% undergoing CABG. The mean treatment duration was
5.5 days in the fondaparinux group and 5.2 days in the enoxaparin group. If PCI was performed,
patients received either intravenous fondaparinux (fondaparinux patients) or weight adjusted
intravenous UFH (enoxaparin patients) as adjunctive therapy, dependent on the timing of the last
subcutaneous dose and planned use of GP IIb/IIIa inhibitor. The mean age of the patients was 67
years, and approximately 60% were at least 65 years old. Approximately 40% and 17% of patients had
23
mild (creatinine clearance ≥50 to <80 ml/min) or moderate (creatinine clearance ≥30 to <50 ml/min)
renal impairment, respectively.
The primary adjudicated endpoint was a composite of death, myocardial infarction (MI) and refractory
ischaemia (RI) within 9 days of randomisation. Of the patients in the fondaparinux group, 5.8%
experienced an event by Day 9 compared to 5.7% for enoxaparin-treated patients (hazard ratio 1.01,
95% CI, 0.90, 1.13, one-sided non-inferiority p value = 0.003).
By Day 30, the incidence of all cause mortality was significantly reduced from 3.5% on enoxaparin to
2.9% on fondaparinux (hazard ratio 0.83, 95% CI, 0.71;0.97, p = 0.02). The effects on the incidence of
MI and RI were not statistically different between the fondaparinux and enoxaparin treatment groups.
At Day 9 the incidence of major bleeding on fondaparinux and enoxaparin was 2.1% and 4.1%,
respectively (hazard ratio 0.52, 95% CI, 0.44;0.61, p < 0.001).
The efficacy findings and results on major bleeding were consistent across prespecified subgroups
such as elderly, renally impaired patients, type of concomitant platelet aggregation inhibitors (aspirin,
thienopyridines or GP IIb/IIIa inhibitors).
In the subgroup of patients treated with fondaparinux or enoxaparin who underwent PCI, 8.8% and
8.2% of patients respectively, experience death/MI/RI within 9 days of randomisation (hazard ratio
1.08, 95% CI, 0.92;1.27). In this subgroup, the incidence of major bleeding on fondaparinux and
enoxaparin at Day 9 was 2.2% and 5.0% respectively (hazard ratio 0.43, 95% CI, 0.33;0.57).
Treatment of ST segment elevation myocardial infarction (STEMI)
OASIS 6 was a double blind, randomised study assessing the safety and efficacy of fondaparinux 2.5
mg once daily, versus usual care (placebo (47%) or UFH (53%) in approximately 12,000 patients with
STEMI. All patients received standard treatments for STEMI, including primary PCI (31%),
thrombolytics (45%) or no reperfusion (24%). Of the patients treated with a thrombolytic, 84% were
treated with a non-fibrin specific agent (primarily streptokinase). The mean treatment duration was
6.2 days on fondaparinux. The mean age of the patients was 61 years, and approximately 40% were at
least 65 years old. Approximately 40% and 14% of patients had mild (creatinine clearance ≥50 to <80
ml/min) or moderate (creatinine clearance ≥30 to <50 ml/min) renal impairment, respectively.
The primary adjudicated endpoint was a composite of death and recurrent MI (re-MI) within 30 days
of randomisation. The incidence of death/re-MI at Day 30 was significantly reduced from 11.1% for
the control group to 9.7% for the fondaparinux group (hazard ratio 0.86, 95% CI, 0.77, 0.96, p =
0.008). In the predefined stratum comparing fondaparinux to placebo (i.e patients treated with non-
fibrin specific lytics (77.3%), no reperfusion (22%), fibrin-specific lytics (0.3%), primary PCI (0.4%),
the incidence of death/re-MI at Day 30 was significantly reduced from 14.0% on placebo to 11.3%
(hazard ratio 0.80, 95% CI, 0.69, 0.93, p = 0.003). In the predefined stratum comparing fondaparinux
to UFH (patients treated with primary PCI (58.5%), fibrin-specific lytics (13%), non-fibrin-specific
lytics (2.6%) and no reperfusion (25.9%), the effects of fondaparinux and UFH on the incidence of
death/re-MI at Day 30 were not statistically different: respectively, 8.3% vs 8.7% (hazard ratio 0.94,
95% CI, 0.79, 1.11 p = 0.460). However, in this stratum, in the subgroup of indicated population
undergoing thrombolysis or no reperfusion (i.e patients not undergoing primary PCI), the incidence of
death/re-MI at Day 30 was significantly reduced from 14.3% on UFH to 11.5% with fondaparinux
(hazard ratio 0.79, 95% CI, 0.64, 0.98, p = 0.03).
The incidence of all cause mortality at Day 30 was also significantly reduced from 8.9% for the
control group to 7.8% in the fondaparinux group (hazard ratio 0.87, 95% CI, 0.77;0.98, p = 0.02). The
difference in mortality was statistically significant in stratum 1 (placebo comparator) but not in
stratum 2 (UFH comparator). The mortality benefit shown in the fondaparinux group was maintained
until the end of follow-up at Day 180.
In patients who were revascularised with a thrombolytic, fondaparinux significantly reduced the
incidence of death/re-MI at Day 30 from 13.6% for the control group to 10.9% (hazard ratio 0.79,
24
95%CI, 0.68;0.93, p = 0.003). Among patients initially not reperfused, the incidence of death/re-MI at
Day 30 was significantly reduced from 15% for the control group to 12.1% for the fondaparinux group
(hazard ratio 0.79, 95% CI, 0.65;0.97, p = 0.023). In patients treated with primary PCI, the incidence
of death/re-MI at Day 30 was not statistically different between the two groups [6.0% in fondaparinux
group vs 4.8% in the control group; hazard ratio 1.26, 95% CI, 0.96, 1.66].
By Day 9, 1.1% of patients treated with fondaparinux and 1.4% of control patients experienced a
severe haemorrhage. In patients given a thrombolytic, severe haemorrhage occurred in 1.3% of the
fondaparinux patients and in 2.0% of controls. In patients initially not reperfused, the incidence of
severe haemorrhage was 1.2% for fondaparinux vs 1.5% for controls. For patients receiving primary
PCI, the incidence of severe haemorrhage was 1.0% for fondaparinux and 0.4% for controls.
The efficacy findings and results on severe haemorrhage were consistent across prespecified
subgroups such as elderly, renally impaired patients, type of concomitant platelet aggregation
inhibitors (aspirin, thienopyridines).
Treatment of patients with acute symptomatic spontaneous superficial-vein thrombosis without
concomitant Deep-Vein Thrombosis (DVT)
A randomized, double blind, clinical trial (CALISTO) included 3002 patients with acute symptomatic
isolated, spontaneous superficial-vein thrombosis of the lower limbs, at least 5 cm long, confirmed by
compression ultrasonography.
Patients were not included if they had concomitant DVT or superficial-
vein thrombosis
within 3 cm of the sapheno-femoral junction. Patients were excluded if they had
severe hepatic impairment, severe renal impairment (creatinine clearance <30ml/min), low body
weight (<50kg), active cancer, symptomatic PE or a recent history of DVT/PE (<6 months) or
superficial-vein thrombosis (<90 days), or superficial-vein thrombosis associated with sclerotherapy or
a complication of an IV line, or they were at high risk of bleeding.
Patients were randomized to receive fondaparinux 2.5 mg once daily or placebo for 45 days in
addition to elastic stockings, analgesic and/or topical NSAIDS anti-inflammatory drugs. Follow-up
continued up to Day 77. The study population was 64% female, with a median age of 58 years, 4.4%
had a creatinine clearance <50ml/min.
The primary efficacy outcome, a composite of symptomatic PE, symptomatic DVT, symptomatic
superficial-vein thrombosis extension, symptomatic superficial-vein thrombosis reoccurrence, or
Death up to Day 47, was significantly reduced from 5.9% in placebo patients to 0.9% in those
receiving fondaparinux 2.5 mg (relative risk reduction: 85.2%; 95% CIs, 73.7% to 91.7% [p<0.001]).
The incidence of each thromboembolic component of the primary outcome was also significantly
reduced in fondaparinux patients as follows: symptomatic PE [0 (0%) vs 5 (0.3%) (p=0.031)],
symptomatic DVT [3 (0.2%) vs 18 (1.2%); relative risk reduction 83.4% (p<0.001)], symptomatic
superficial-vein thrombosis extension [4 (0.3%) vs 51 (3.4%); relative risk reduction 92.2%
(p<0.001)], symptomatic superficial-vein thrombosis reoccurrence [5 (0.3%) vs 24 (1.6%); relative
risk reduction 79.2% (p<0.001)].
The mortality rates were low and similar between the treatments groups with
2 (0.1%) deaths in
the fondaparinux group versus 1 (0.1%) death in the placebo group.
Efficacy was maintained up to Day 77 and was consistent across all predefined subgroups including
patients with varicose veins and patients with superficial-vein thrombosis located below the knee.
Major bleeding during treatment occurred in 1 (0.1%) fondaparinux patient and in 1 (0.1%) placebo
patient. Clinically relevant non major bleeding occurred in 5 (0.3%) fondaparinux patients and 8
(0.5%) placebo patients.
25
5.2 Pharmacokinetic properties
Absorption
After subcutaneous dosing, fondaparinux is completely and rapidly absorbed (absolute bioavailability
100%). Following a single subcutaneous injection of fondaparinux
2.5 mg to young healthy subjects,
peak plasma concentration (mean C
max
= 0.34 mg/l) is obtained 2 hours post-dosing. Plasma
concentrations of half the mean C
max
values are reached 25 minutes post-dosing.
In elderly healthy subjects, pharmacokinetics of fondaparinux are linear in the range of 2 to 8 mg by
subcutaneous route. Following once daily subcutaneous dosing, steady state of plasma levels is
obtained after 3 to 4 days with a 1.3-fold increase in C
max
and AUC.
Mean (CV%) steady state pharmacokinetic parameters estimates of fondaparinux in patients
undergoing hip replacement surgery receiving fondaparinux
2.5 mg once daily are: C
max
(mg/l) - 0.39
(31%), T
max
(h) - 2.8 (18%) and C
min
(mg/l) -0.14 (56%). In hip fracture patients, associated with their
increased age, fondaparinux steady state plasma concentrations are: C
max
(mg/l) - 0.50 (32%),
C
min
(mg/l) - 0.19 (58%).
Distribution
The distribution volume of fondaparinux is limited (7-11 litres).
In vitro
, fondaparinux is highly and
specifically bound to antithrombin protein with a dose-dependant plasma concentration binding
(98.6% to 97.0% in the concentration range from 0.5 to 2 mg/l). Fondaparinux does not bind
significantly to other plasma proteins, including platelet factor 4 (PF4).
Since fondaparinux does not bind significantly to plasma proteins other than ATIII, no interaction
with other medicinal products by protein binding displacement are expected.
Biotransformation
Although not fully evaluated, there is no evidence of fondaparinux metabolism and in particular no
evidence for the formation of active metabolites.
Fondaparinux does not inhibit CYP450s (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6,
CYP2E1 or CYP3A4)
in vitro
. Thus, fondaparinux is not expected to interact with other medicinal
products
in vivo
by inhibition of CYP-mediated metabolism.
Excretion/Elimination
The elimination half-life (t
½
) is about 17 hours in healthy young subjects and about 21 hours in healthy
elderly subjects. Fondaparinux is excreted to 64 – 77 % by the kidney as unchanged compound.
Special populations
Paediatric patients
- Fondaparinux
has not been investigated in this population for the prevention of
VTE or for the treatment of superficial vein thrombosis or acute coronary syndrome (ACS).
Elderly patients
- Renal function may decrease with age and thus, the elimination capacity for
fondaparinux may be reduced in elderly.
In patients >75 years undergoing orthopaedic surgery, the
estimated plasma clearance was 1.2 to 1.4 times lower than in patients <65 years.
Renal impairment
- Compared with patients with normal renal function (creatinine
clearance > 80 ml/min), plasma clearance is 1.2 to 1.4 times lower in patients with mild renal
impairment (creatinine clearance 50 to 80 ml/min) and on average 2 times lower in patients with
moderate renal impairment (creatinine clearance 30 to 50 ml/min). In severe renal impairment
(creatinine clearance < 30 ml/min), plasma clearance is approximately 5 times lower than in normal
renal function. Associated terminal half-life values were 29 h in moderate and 72 h in patients with
severe renal impairment.
Gender
- No gender differences were observed after adjustment for body weight.
26
Race
- Pharmacokinetic differences due to race have not been studied prospectively. However, studies
performed in Asian (Japanese) healthy subjects did not reveal a different pharmacokinetic profile
compared to Caucasian healthy subjects. Similarly, no plasma clearance differences were observed
between black and Caucasian patients undergoing orthopaedic surgery.
Body weight
-
Plasma clearance of fondaparinux increases with body weight (9% increase per 10 kg).
Hepatic impairment -
Following a single, subcutaneous dose of fondaparinux in subjects with
moderate hepatic impairment (Child-Pugh Category B), total (i.e., bound and unbound) C
max
and AUC
were decreased by 22% and 39%, respectively, as compared to subjects with normal liver function.
The lower plasma concentrations of fondaparinux were attributed to reduced binding to ATIII
secondary to the lower ATIII plasma concentrations in subjects with hepatic impairment thereby
resulting in increased renal clearance of fondaparinux. Consequently, unbound concentrations of
fondaparinux are expected to be unchanged in patients with mild to moderate hepatic impairment, and
therefore, no dose adjustment is necessary based on pharmacokinetics.
The pharmacokinetics of fondaparinux has not been studied in patients with severe hepatic impairment
(see sections 4.2 and 4.4).
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity, and genotoxicity. Animal studies are insufficient with respect to
effects on toxicity to reproduction because of limited exposure.
6.
6.1 List of excipients
Sodium chloride
Water for injections
Hydrochloric acid
Sodium hydroxide
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3 Shelf life
3 years.
If fondaparinux sodium is added to a 0.9% saline minibag it should ideally be infused immediately,
but can be stored at room temperature for up to 24 hours.
6.4 Special precautions for storage
Do not freeze.
6.5 Nature and contents of container
Type I glass barrel (1 ml) affixed with a 27 gauge x 12.7 mm needle and stoppered with a bromobutyl
or chlorobutyl elastomer plunger stopper.
Arixtra
is available in pack sizes of 2, 7, 10 and 20 pre-filled syringes. There are two types of syringes:
27
•
syringe with a blue plunger and an automatic safety system
•
syringe with blue plunger and a manual safety system.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
The subcutaneous injection is administered in the same way as with a classical syringe. Intravenous
administration should be through an existing intravenous line either directly or using a small volume
(25 or 50ml) 0.9% saline minibag.
Parenteral solutions should be inspected visually for particulate matter and discoloration prior to
administration.
Instruction on self-administration by subcutaneous injection is included in the Package Leaflet.
The needle protection system of the Arixtra pre-filled syringes have been designed with a safety
system to protect from needle stick injuries following injection.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Glaxo Group Ltd
Greenford
Middlesex
UB6 0NN
United Kingdom
8.
EU/1/02/206/001-004
EU/1/02/206/021
EU/1/02/206/022
EU/1/02/206/023
9.
Date of first authorisation: 21 March 2002
Date of latest renewal: 21 March 2007
Detailed information on this medicinal product is available on the website of the European Medicines
Agency http://www.ema.europa.eu
28
1.
Arixtra 5 mg/0.4 ml solution for injection, pre-filled syringe.
2.
Each pre-filled syringe contains 5 mg of fondaparinux sodium in 0.4 ml solution for injection.
Excipient(s): Contains less than 1 mmol of sodium (23 mg) per dose, and therefore is essentially
sodium free.
For a full list of excipients, see section 6.1.
3.
Solution for injection.
The solution is a clear and colourless to slightly yellow liquid.
4.
Treatment of adults with acute Deep Vein Thrombosis (DVT) and treatment of acute Pulmonary
Embolism (PE), except in haemodynamically unstable patients or patients who require thrombolysis or
pulmonary embolectomy
.
Posology
The recommended dose of fondaparinux is 7.5 mg (patients with body weight ≥ 50, ≤ 100kg) once
daily administered by subcutaneous injection. For patients with body weight < 50 kg, the
recommended dose is 5 mg. For patients with body weight > 100 kg, the recommended dose is 10 mg.
Treatment should be continued for at least 5 days and until adequate oral anticoagulation is established
(International Normalised Ratio 2 to 3). Concomitant oral anticoagulation treatment should be initiated
as soon as possible and usually within 72 hours. The average duration of administration in clinical
trials was 7 days and the clinical experience from treatment beyond 10 days is limited.
Special populations
Elderly patients
- No dosing adjustment is necessary.
In patients ≥75 years, fondaparinux should be
used with care, as renal function decreases with age (see section 4.4).
Renal impairment -
Fondaparinux should be used with caution in patients with moderate renal
impairment (see section 4.4).
There is no experience in the subgroup of patients with
both
high body weight (>100 kg) and moderate
renal impairment (creatinine clearance 30-50 ml/min). In this subgroup, after an initial 10 mg daily
dose, a reduction of the daily dose to 7.5 mg may be considered, based on pharmacokinetic modelling
(see section 4.4).
Fondaparinux should not be used in patients with severe renal impairment (creatinine clearance < 30
ml/min) (see section 4.3).
29
Hepatic impairment
- No dosing adjustment is necessary in patients with either mild or moderate
hepatic impairment. In patients with severe hepatic impairment, fondaparinux should be used with
care as this patient group has not been studied (see sections 4.4 and 5.2).
Paediatric population -
Fondaparinux is not recommended for use in children below 17 years of age
due to a lack of data on safety and efficacy (see sections 5.1 and 5.2).
Method of administration
Fondaparinux is administered by deep subcutaneous injection while the patient is lying down. Sites of
administration should alternate between the left and the right anterolateral and left and right
posterolateral abdominal wall. To avoid the loss of medicinal product when using the pre-filled
syringe do not expel the air bubble from the syringe before the injection. The whole length of the
needle should be inserted perpendicularly into a skin fold held between the thumb and the forefinger;
the skin fold should be held throughout the injection.
For additional instructions for use and handling and disposal see section 6.6.
-
hypersensitivity to the active substance or to any of the excipients
-
active clinically significant bleeding
-
severe renal impairment defined by creatinine clearance < 30 ml/min.
Fondaparinux is intended for subcutaneous use only. Do not administer intramuscularly
.
There is limited experience from treatment with fondaparinux in haemodynamically unstable patients
and no experience in patients requiring thrombolysis, embolectomy or insertion of a vena cava filter.
Haemorrhage
Fondaparinux
should be used with caution in patients who have an increased risk of haemorrhage,
such as those with congenital or acquired bleeding disorders (e.g. platelet count <50,000/mm
3
), active
ulcerative gastrointestinal disease and recent intracranial haemorrhage or shortly after brain, spinal or
ophthalmic surgery and in special patient groups as outlined below.
As for other anticoagulants, fondaparinux should be used with caution in patients who have undergone
recent surgery (<3 days) and only once surgical haemostasis has been established
.
Agents that may enhance the risk of haemorrhage should not be administered concomitantly with
fondaparinux. These agents include desirudin, fibrinolytic agents, GP IIb/IIIa receptor antagonists,
heparin, heparinoids, or Low Molecular Weight Heparin (LMWH). During treatment of VTE,
concomitant therapy with vitamin K antagonist should be administered in accordance with the
information of Section 4.5. Other antiplatelet medicinal products (acetylsalicylic acid, dipyridamole,
sulfinpyrazone, ticlopidine or clopidogrel), and NSAIDs should be used with caution. If co-
administration is essential, close monitoring is necessary.
Spinal / Epidural anaesthesia
In patients receiving fondaparinux for treatment of VTE rather than prophylaxis, spinal/epidural
anaesthesia in case of surgical procedures should not be used.
Elderly patients
The elderly population is at increased risk of bleeding. As renal function generally decreases with age,
elderly patients may show reduced elimination and increased exposure of fondaparinux (see section
5.2). Incidences of bleeding events in patients receiving the recommended regimen in the treatment of
DVT or PE and aged <65 years, 65-75 and >75 years were 3.0 %, 4.5 % and 6.5 %, respectively. The
30
-
acute bacterial endocarditis
corresponding incidences in patients receiving the recommended regimen of enoxaparin in the
treatment of DVT were 2.5%, 3.6% and 8.3% respectively, while the incidences in patients receiving
the recommended regimen of UFH in the treatment of PE were 5.5%, 6.6% and 7.4%, respectively.
Fondaparinux should be used
with caution in elderly patients (see section 4.2).
Low body weight
Clinical experience is limited in patients with body weight <50 kg. Fondaparinux should be used with
caution at a daily dose of 5 mg in this population (see sections 4.2 and 5.2).
Renal impairment
The risk of bleeding increases with increasing renal impairment. Fondaparinux is known to be
excreted mainly by the kidney. Incidences of bleeding events in patients receiving the recommended
regimen in the treatment of DVT or PE with normal renal function, mild renal impairment, moderate
renal impairment and severe renal impairment were 3.0 % (34/1,132), 4.4 % (32/733), 6.6% (21/318),
and 14.5 % (8/55) respectively. The corresponding incidences in patients receiving the recommended
regimen of enoxaparin in the treatment of DVT were 2.3% (13/559), 4.6% (17/368), 9.7% (14/145)
and 11.1% (2/18) respectively, and in patients receiving the recommended regimen of unfractionated
heparin in the treatment of PE were 6.9% (36/523), 3.1% (11/352), 11.1% (18/162) and 10.7% (3/28),
respectively.
Fondaparinux is contra-indicated in severe renal impairment (creatinine clearance <30 ml/min) and
should be used with caution in patients with moderate renal impairment (creatinine clearance 30-50
ml/min). The duration of treatment should not exceed that evaluated during clinical trial (mean 7 days)
(see sections 4.2, 4.3 and 5.2).
There is no experience in the subgroup of patients with both high body weight (>100 kg) and moderate
renal impairment (creatinine clearance 30-50 ml/min). Fondaparinux should be used with care in these
patients. After an initial 10 mg daily dose, a reduction of the daily dose to 7.5 mg may be considered,
based on pharmacokinetic modelling (see section 4.2).
Severe hepatic impairment
The use of fondaparinux should be considered with caution because of an increased risk of bleeding
due to a deficiency of coagulation factors in patients with severe hepatic impairment (see section 4.2).
Patients with Heparin Induced Thrombocytopenia
Fondaparinux should be used with caution in patients with a history of HIT. The efficacy and safety of
fondaparinux have not been formally studied in patients with HIT type II. Fondaparinux does not bind
to platelet factor 4 and does not cross-react with sera from patients with Heparin Induced
Thrombocytopenia (HIT) type II
.
However,
rare spontaneous reports of HIT in patients treated with
fondaparinux have been received. To date a causal association between treatment with fondaparinux
and the occurrence of HIT has not been established.
Bleeding risk is increased with concomitant administration of fondaparinux and agents that may
enhance the risk of haemorrhage (see section 4.4).
In clinical studies performed with fondaparinux, oral anticoagulants (warfarin) did not interact with
the pharmacokinetics of fondaparinux; at the 10 mg dose used in the interaction studies, fondaparinux
did not influence the anticoagulation monitoring (INR) activity of warfarin.
Platelet inhibitors (acetylsalicylic acid), NSAIDs (piroxicam) and digoxin did not interact with the
pharmacokinetics of fondaparinux. At the 10 mg dose used in the interaction studies, fondaparinux did
not influence the bleeding time under acetylsalicylic acid or piroxicam treatment, nor the
pharmacokinetics of digoxin at steady state.
31
Pregnancy
No clinical data on exposed pregnancies are available. Animal studies are insufficient with respect to
effects on pregnancy, embryo/foetal development, parturition and postnatal development because of
limited exposure. Fondaparinux should not be prescribed to pregnant women unless clearly necessary.
Breastfeeding
Fondaparinux is excreted in rat milk but it is not known whether fondaparinux is excreted in human
milk. Breastfeeding is not recommended during treatment with fondaparinux. Oral absorption by the
child is however unlikely.
Fertility
There are no data available on the effect of fondaparinux on human fertility. Animal studies do not
show any effect on fertility.
No studies on the effect on the ability to drive and to use machines have been performed.
The most commonly reported serious adverse reactions reported with fondaparinux are bleeding
complications (various sites including rare cases of intracranial/ intracerebral and retroperitoneal
bleedings) and anaemia. Fondaparinux should be used with caution in patients who have an increased
risk of haemorrhage (see section 4.4).
The safety of
fondaparinux has been evaluated in 2,517 patients treated for Venous Thrombo-
Embolism and treated with fondaparinux for an average of 7 days. The most common adverse
reactions were bleeding complications (see section 4.4).
The adverse reactions reported by the investigator as at least possibly related to fondaparinux are
presented within each frequency grouping (very common ≥ 1/10; common: ≥1/100 to < 1/10;
uncommon: ≥ 1/1,000 to < 1/100; rare: ≥ 1/10,000 to <1/1,000; very rare <1/10,000) and system organ
class by decreasing order of seriousness.
32
System organ class
MedDRA
Adverse reactions in patients treated for VTE
1
Blood and lymphatic
system disorders
Common:
bleeding (gastrointestinal, haematuria,
haematoma, epistaxis, haemoptysis, utero-vaginal
haemorrhage, haemarthrosis, ocular, purpura,
bruise)
Uncommon:
anaemia, thrombocytopaenia
Rare:
other bleeding (hepatic, retroperitoneal,
intracranial/intracerebral), thrombocythaemia
Immune system
disorders
Rare:
allergic reaction
Metabolism and
nutrition disorders
Rare:
non-protein-nitrogen (Npn)
2
increased
Nervous system
disorders
Uncommon
: headache
Rare:
dizziness
Gastrointestinal
disorders
Uncommon:
nausea, vomiting
Hepatobiliary disorders Uncommon
: abnormal liver function
Skin and subcutaneous
tissue disorders
Rare:
rash erythematous
General disorders and
administration site
conditions
Uncommon:
pain, oedema,
Rare:
reaction at injection site
(1) Isolated AEs have not been considered except if they were medically relevant.
(2) Npn stands for non-protein-nitrogen such as urea, uric acid, amino acid, etc.
Fondaparinux doses above the recommended regimen
may lead to an increased risk of bleeding. There
is no known antidote to fondaparinux.
for the primary cause. Initiation of appropriate therapy such as surgical haemostasis, blood
replacements, fresh plasma transfusion, plasmapheresis should be considered.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antithrombotic agents.
ATC code: B01AX05
33












Pharmacodynamic effects
Fondaparinux is a synthetic and selective inhibitor of activated Factor X (Xa). The antithrombotic
activity of fondaparinux is the result of antithrombin III (antithrombin) mediated selective inhibition
of Factor Xa. By binding selectively to antithrombin, fondaparinux potentiates (about 300 times) the
innate neutralization of Factor Xa by antithrombin. Neutralisation of Factor Xa interrupts the blood
coagulation cascade and inhibits both thrombin formation and thrombus development. Fondaparinux
does not inactivate thrombin (activated Factor II) and has no effects on platelets.
At the doses used for treatment, fondaparinux does not
,
to a clinically relevant extent, affect routine
coagulation tests such as activated partial thromboplastin time (aPTT), activated clotting time (ACT)
or prothrombin time (PT)/International Normalised Ratio (INR) tests in plasma nor bleeding time or
fibrinolytic activity. However, rare spontaneous reports of aPTT prolongation have been received. At
higher doses, moderate changes in aPTT can occur. At the 10 mg dose used in interaction studies,
fondaparinux did not significantly
influence the anticoagulation activity (INR) of warfarin.
Fondaparinux does not cross-react with sera from patients with heparin-induced thrombocytopaenia.
Clinical studies
The fondaparinux clinical program in treatment of Venous Thromboembolism was designed to
demonstrate the efficacy of fondaparinux for the treatment of deep vein thrombosis (DVT) and
pulmonary embolism (PE). Over 4,874 patients were studied in controlled Phase II and III clinical
studies.
Treatment of Deep Venous Thrombosis
In a randomised, double-blind, clinical trial in patients with a confirmed diagnosis of acute
symptomatic DVT, fondaparinux 5 mg (body weight < 50 kg), 7.5 mg (body weight ≥ 50 kg, ≤ 100
kg) or 10 mg (body weight >100 kg) SC once daily was compared to enoxaparin sodium 1 mg/kg SC
twice daily. A total of 2,192 patients were treated; for both groups, patients were treated for at least 5
days and up to 26 days (mean 7 days). Both treatment groups received Vitamin K antagonist therapy
usually initiated within 72 hours after the first study drug administration and continued for 90 ± 7
days, with regular dose adjustments to achieve an INR of 2-3. The primary efficacy endpoint was the
composite of confirmed symptomatic recurrent non-fatal VTE and fatal VTE reported up to Day 97.
Treatment with fondaparinux was demonstrated to be non-inferior to enoxaparin (VTE rates 3.9% and
4.1%, respectively).
Major bleeding during the initial treatment period was observed in 1.1% of fondaparinux patients,
compared to 1.2% with enoxaparin.
Treatment of Pulmonary Embolism
A randomised, open-label, clinical trial was conducted in patients with acute symptomatic PE. The
diagnosis was confirmed by objective testing (lung scan, pulmonary angiography or spiral CT scan).
Patients who required thrombolysis or embolectomy or vena cava filter were excluded. Randomised
patients could have been pre-treated with UFH during the screening phase but patients treated for
more than 24 hours with therapeutic dose of anticoagulant or with uncontrolled hypertension were
excluded. Fondaparinux 5 mg (body weight < 50 kg), 7.5 mg (body weight ≥ 50kg, ≤ 100 kg) or 10
mg (body weight >100 kg) SC once daily was compared to unfractionated heparin IV bolus (5,000 IU)
followed by a continuous IV infusion adjusted to maintain 1.5–2.5 times aPTT control value. A total
of 2,184 patients were treated; for both groups, patients were treated for at least 5 days and up to 22
days (mean 7 days). Both treatment groups received Vitamin K antagonist therapy usually initiated
within 72 hours after the first study drug administration and continued for 90 ± 7 days, with regular
dose adjustments to achieve an INR of 2-3. The primary efficacy endpoint was the composite of
confirmed symptomatic recurrent non-fatal VTE and fatal VTE reported up to Day 97. Treatment with
fondaparinux was demonstrated to be non-inferior to unfractionated heparin (VTE rates 3.8% and
5.0%, respectively).
34
Major bleeding during the initial treatment period was observed in 1.3% of fondaparinux patients,
compared to 1.1% with unfractionated heparin.
A pilot dose-finding and pharmacokinetic study of fondaparinux in children with deep vein
thrombosis
In an open-label study, 24 paediatric patients (n=10, age 1 to ≤ 5 years weight range 8-20 kg; n=7, age
6 to ≤ 12 years weight range 17-47 kg and n=7 age 13 to ≤ 18 years weight range 47-130 kg)
diagnosed with venous thrombosis at study entry were administered fondaparinux. The majority of
patients were Hispanic (67%) and 58% were male. Fondaparinux was administered at an initial dose of
0.1 mg/kg subcutaneously once daily and dosing was adjusted to achieve peak fondaparinux sodium
concentrations of 0.5 to 1 mg/L after 4 hours. The median duration of treatment in this study was 3.5
days. The majority of patients (88%) achieved target fondaparinux concentrations at 4 hours after the
first dose of fondaparinux. Two patients had reports of bleeding during the study. One experienced
hypertensive encephalopathy accompanied by intracranial bleeding on day 5 of therapy resulting in
fondaparinux discontinuation. Minor gastrointestinal bleeding was reported in another patient on day 5
of therapy which resulted in temporary discontinuation of fondaparinux. No conclusion can be drawn
with regard to clinical efficacy in this uncontrolled study.
5.2 Pharmacokinetic properties
The pharmacokinetics of fondaparinux sodium are derived from fondaparinux plasma concentrations
quantified via anti factor Xa activity. Only fondaparinux can be used to calibrate the anti-Xa assay (the
international standards of heparin or LMWH are not appropriate for this use). As a result, the
concentration of fondaparinux is expressed as milligrams (mg).
Absorption
After subcutaneous dosing, fondaparinux is completely and rapidly absorbed (absolute bioavailability
100%). Following a single subcutaneous injection of fondaparinux
2.5 mg to young healthy subjects,
peak plasma concentration (mean C
max
= 0.34 mg/l) is obtained 2 hours post-dosing. Plasma
concentrations of half the mean C
max
values are reached 25 minutes post-dosing.
In elderly healthy subjects, pharmacokinetics of fondaparinux is linear in the range of 2 to 8 mg by
subcutaneous route. Following once daily dosing, steady state of plasma levels is obtained after 3 to 4
days with a 1.3-fold increase in C
max
and AUC.
Mean (CV%) steady state pharmacokinetic parameters estimates of fondaparinux in patients
undergoing hip replacement surgery receiving fondaparinux
2.5 mg once daily are: C
max
(mg/l) - 0.39
(31%), T
max
(h) - 2.8 (18%) and C
min
(mg/l) -0.14 (56%). In hip fracture patients, associated with their
increased age, fondaparinux steady state plasma concentrations are: C
max
(mg/l) - 0.50 (32%),
C
min
(mg/l) - 0.19 (58%).
In DVT and PE treatment, patients receiving fondaparinux 5 mg (body weight <50 kg), 7.5 mg (body
weight 50-100 kg inclusive) and 10 mg (body weight >100 kg) once daily, the body weight-adjusted
doses provide similar exposure across all body weight categories. The mean (CV%) steady state
pharmacokinetic parameters estimates of fondaparinux in patients with VTE receiving the
fondaparinux proposed dose regimen once daily are: C
max
(mg/l) - 1.41 (23 %), T
max
(h) – 2.4 (8%) and
C
min
(mg/l) -0.52 (45 %). The associated 5th and 95th percentiles are, respectively, 0.97 and 1.92 for
C
max
(mg/l), and 0.24 and 0.95 for C
min
(mg/l).
Distribution
The distribution volume of fondaparinux is limited (7-11 litres).
In vitro
, fondaparinux is highly and
specifically bound to antithrombin protein with a dose-dependant plasma concentration binding
(98.6% to 97.0% in the concentration range from 0.5 to 2 mg/l). Fondaparinux does not bind
significantly to other plasma proteins, including platelet factor 4 (PF4).
Since fondaparinux does not bind significantly to plasma proteins other than antithrombin, no
interaction with other medicinal products by protein binding displacement are expected.
35
Biotransformation
Although not fully evaluated, there is no evidence of fondaparinux metabolism and in particular no
evidence for the formation of active metabolites.
Fondaparinux does not inhibit CYP450s (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6,
CYP2E1 or CYP3A4)
in vitro
. Thus, fondaparinux is not expected to interact with other medicinal
products
in vivo
by inhibition of CYP-mediated metabolism.
Excretion/Elimination
The elimination half-life (t
½
) is about 17 hours in healthy young subjects and about 21 hours in healthy
elderly subjects. Fondaparinux is excreted to 64 – 77 % by the kidney as unchanged compound.
Special populations
Paediatric patients
- Limited data are available in paediatric patients (see section 5.1).
Elderly patients
- Renal function may decrease with age and thus, the elimination capacity for
fondaparinux may be reduced in elderly.
In patients >75 years undergoing orthopaedic surgery and
receiving fondaparinux 2.5 mg once daily, the estimated plasma clearance was 1.2 to 1.4 times lower
than in patients <65 years. A similar pattern is observed in DVT and PE treatment patients.
Renal impairment
- Compared with patients with normal renal function (creatinine clearance
> 80 ml/min) undergoing orthopaedic surgery and receiving fondaparinux
2.5 mg once daily, plasma
clearance is 1.2 to 1.4 times lower in patients with mild renal impairment (creatinine clearance 50 to
80 ml/min) and on average 2 times lower in patients with moderate renal impairment (creatinine
clearance 30 to 50 ml/min). In severe renal impairment (creatinine clearance <30 ml/min), plasma
clearance is approximately 5 times lower than in normal renal function. Associated terminal half-life
values were 29 h in moderate and 72 h in patients with severe renal impairment. A similar pattern is
observed in DVT and PE treatment patients.
Body weight
-
Plasma clearance of fondaparinux increases with body weight (9% increase per 10 kg).
Gender
- No gender differences were observed after adjustment for body weight.
Race
- Pharmacokinetic differences due to race have not been studied prospectively. However, studies
performed in Asian (Japanese) healthy subjects did not reveal a different pharmacokinetic profile
compared to Caucasian healthy subjects. Similarly, no plasma clearance differences were observed
between black and Caucasian patients undergoing orthopaedic surgery.
Hepatic impairment -
Following a single, subcutaneous dose of fondaparinux in subjects with
moderate hepatic impairment (Child-Pugh Category B), total (i.e., bound and unbound) C
max
and AUC
were decreased by 22% and 39%, respectively, as compared to subjects with normal liver function.
The lower plasma concentrations of fondaparinux were attributed to reduced binding to ATIII
secondary to the lower ATIII plasma concentrations in subjects with hepatic impairment thereby
resulting in increased renal clearance of fondaparinux. Consequently, unbound concentrations of
fondaparinux are expected to be unchanged in patients with mild to moderate hepatic impairment, and
therefore, no dose adjustment is necessary based on pharmacokinetics.
The pharmacokinetics of fondaparinux has not been studied in patients with severe hepatic impairment
(see sections 4.2 and 4.4).
36
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology and genotoxicity. The repeated dose and reproduction toxicity studies did not reveal
any special risk but did not provide adequate documentation of safety margins due to limited exposure
in the animal species
.
6.
6.1 List of excipients
Sodium chloride
Water for injections
Hydrochloric acid
Sodium hydroxide
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3 Shelf life
3 years
6.4 Special precautions for storage
Do not freeze.
6.5 Nature and contents of container
Type I glass barrel (1 ml) affixed with a 27 gauge x 12.7 mm needle and stoppered with a chlorobutyl
elastomer plunger stopper.
Arixtra 5 mg/0.4 ml
is available in pack sizes of 2, 7, 10 and 20 pre-filled syringes. There are two
types of syringes:
•
syringe with a orange plunger and an automatic safety system
•
syringe with orange plunger and a manual safety system.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
The subcutaneous injection is administered in the same way as with a classical syringe.
Parenteral solutions should be inspected visually for particulate matter and discoloration prior to
administration.
Instruction for self-administration is mentioned in the Package Leaflet.
The Arixtra pre-filled syringes have been designed with a needle protection system to prevent needle
stick injuries following injection.
Any unused product or waste material should be disposed of in accordance with local requirements.
This medicinal product is for single use only.
37
7.
Glaxo Group Ltd
Greenford
Middlesex
UB6 0NN
United Kingdom
8.
EU/1/02/206/009-011, 018
EU/1/02/206/027
EU/1/02/206/028
EU/1/02/206/033
9.
Date of first authorisation: 21 March 2002
Date of latest renewal: 21 March 2007
Detailed information on this medicinal product is available on the website of the European Medicines
Agency http://www.ema.europa.eu
38
1.
FURTHER INFORMATION
What Arixtra contains
The active substance is:
•
5 mg fondaparinux sodium in 0.4 ml solution for injection
•
7.5 mg fondaparinux sodium in 0.6 ml solution for injection
•
10 mg fondaparinux sodium in 0.8 ml solution for injection
The other ingredient(s) are sodium chloride, water for injections, and hydrochloric acid and/or sodium
hydroxide to adjust the pH.
Arixtra does not contain any animal products.
What Arixtra looks like and contents of the pack
Arixtra is a clear and colourless to slightly yellow solution for injection. It is supplied in a pre-filled
syringe fitted with a safety system to help prevent needle stick injuries after use.
It is available in packs of 2, 7, 10 and 20 pre-filled syringes (not all pack sizes may be marketed).
Marketing Authorisation Holder and Manufacturer
Marketing Authorization Holder:
Glaxo Group Ltd, Greenford, Middlesex, UB6 0NN, United Kingdom
Manufacturer:
Glaxo Wellcome Production, 1 rue de l'Abbaye, F-76960 Notre Dame de Bondeville, France.
This leaflet was last approved in
Detailed information on this medicine is available on the European Medicines Agency web site:
http://www.ema.europa.eu
100
For any information about this medicinal product, please contact the local representative of the
Marketing Authorisation Holder.
België/Belgique/Belgien
GlaxoSmithKline s.a./n.v.
Tél/Tel: + 32 (0)2 656 21 11
Luxembourg/Luxemburg
GlaxoSmithKline s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0)2 656 21 11
България
ГлаксоСмитКлайн ЕООД
Teл.: + 359 2 953 10 34
Magyarország
GlaxoSmithKline Kft.
Tel.: + 36 1 225 5300
Česká republika
GlaxoSmithKline s.r.o.
Tel: + 420 222 001 111
gsk.czmail@gsk.com
Malta
GlaxoSmithKline Malta
Tel: + 356 21 238131
Danmark
GlaxoSmithKline Pharma A/S
Tlf: + 45 36 35 91 00
dk-info@gsk.com
Nederland
GlaxoSmithKline BV
Tel: + 31 (0)30 6938100
nlinfo@gsk.com
Deutschland
GlaxoSmithKline GmbH & Co. KG
Tel.: + 49 (0)89 36044 8701
produkt.info@gsk.com
Norge
GlaxoSmithKline AS
Tlf: + 47 22 70 20 00
firmapost@gsk.no
Eesti
GlaxoSmithKline Eesti OÜ
Tel: + 372 6676 900
Österreich
GlaxoSmithKline Pharma GmbH
Tel: + 43 (0)1 97075 0
at.info@gsk.com
Ελλάδα
GlaxoSmithKline A.E.B.E.
Τηλ: + 30 210 68 82 100
Polska
GSK Commercial Sp. z o.o.
Tel.: + 48 (0)22 576 9000
España
GlaxoSmithKline, S.A.
Tel: + 34 902 202 700
es-ci@gsk.com
Portugal
GlaxoSmithKline – Produtos Farmacêuticos, Lda.
Tel: + 351 21 412 95 00
FI.PT@gsk.com
France
Laboratoire GlaxoSmithKline
Tél.: + 33 (0)1 39 17 84 44
diam@gsk.com
România
GlaxoSmithKline (GSK) S.R.L.
Tel: + 4021 3028 208
Ireland
GlaxoSmithKline (Ireland) Limited
Tel: + 353 (0)1 4955000
Slovenija
GlaxoSmithKline d.o.o.
Tel: + 386 (0)1 280 25 00
medical.x.si@gsk.com
Ísland
GlaxoSmithKline ehf.
Sími: + 354 530 3700
Slovenská republika
GlaxoSmithKline Slovakia s. r. o.
Tel: + 421 (0)2 48 26 11 11
recepcia.sk@gsk.com
101
Italia
GlaxoSmithKline S.p.A.
Tel: + 39 (0)45 9218 111
Suomi/Finland
GlaxoSmithKline Oy
Puh/Tel: + 358 (0)10 30 30 30
Κύπρος
GlaxoSmithKline (Cyprus) Ltd
Τηλ: + 357 22 39 70 00
Sverige
GlaxoSmithKline AB
Tel: + 46 (0) 8 638 93 00
info.produkt@gsk.com
Latvija
GlaxoSmithKline Latvia SIA
Tel: + 371 67312687
lv-epasts@gsk.com
United Kingdom
GlaxoSmithKline UK
Tel: + 44 (0)800 221441
customercontactuk@gsk.com
Lietuva
GlaxoSmithKline Lietuva UAB
Tel: + 370 5 264 90 00
info.lt@gsk.com
102
Types of safety syringe
There are two types of safety syringes used for Arixtra, designed to protect you from needle stick
injuries following injection. One type of syringe has an
automatic
needle protection system and the
other type has a
manual
needle protection system.
Parts of the syringes:
1
Needle guard
2
Plunger
3
Finger-grip
4
Security sleeve
Picture 1
. Syringe with an
automatic
needle protection system
Syringe with a
manual
needle protection system
Picture 2.
Syringe with a
manual
needle
protection system
Picture 3.
Syringe with a
manual
needle
protection system showing security sleeve being
pulled over needle
AFTER USE
STEP BY STEP GUIDE TO USING ARIXTRA
Instructions for use
These instructions are for both types of syringes (automatic and manual needle protection system).
Where the instruction for a syringe is different this is clearly stated.
1. Wash your hands thoroughly
with soap and water and dry them with a towel.
2. Remove the syringe from the carton and check that:
•
the expiry date has not passed
•
the solution is clear and colourless to slightly yellow and doesn’t contain particles
103





•
the syringe has not been opened or damaged
3.
Sit or lie down in a comfortable position.
Choose a place in the lower abdominal (tummy) area, at least
5 cm below your belly button (picture
A
).
Alternate the left and right side
of the lower abdominal area
at each injection. This will help to reduce the discomfort at the
injection site.
If injecting in the lower abdominal area is not possible, ask
your nurse or doctor for advice.
Picture A
4. Clean the injection area with an alcohol wipe.
5.
Remove the needle guard
, by first twisting it (picture
B1
),
and then pulling it in a straight line away from the body of the
syringe (picture
B2
).
Discard the needle guard.
Important note
•
Do not touch the needle
or allow it to touch any surface
before the injection.
•
It is normal to see a small air bubble in this syringe.
Do
not try to remove this air bubble before making the
injection
- you may lose some of the medicine if you do.
Picture B1
Picture B2
6. Gently pinch the skin that has been cleaned to make a
fold.
Hold the fold between the thumb and the forefinger
during the entire injection (picture
C
).
Picture C
104
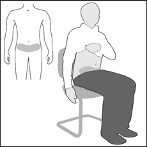



7. Hold the syringe firmly by the finger grip.
Insert the full length of the needle at right angles into the skin
fold (picture
D
).
Picture D
8. Inject ALL of the contents of the syringe by pressing
down on the plunger as far as it goes
(picture
E
).
Syringe automatic system
9. Release the plunger
and the needle will automatically
withdraw from the skin and go back into the security sleeve
where it will be locked permanently (picture
F
).
Picture E
Picture F
Syringe manual system
9.
After the injection hold the syringe in one hand by gripping the security sleeve, use
the other hand to hold the finger grip and pull firmly back. This unlocks the sleeve.
Slide the sleeve up the body of the syringe until it locks into position over the needle.
This is shown in Picture
3
at the beginning of these instructions
Do not dispose of the used syringe in the household waste
. Dispose of it as your doctor or
pharmacist has instructed.
105



Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/arixtra.html
Copyright © 1995-2021 ITA all rights reserved.