










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Eporatio 1,000 IU/0.5 ml solution for injection in pre-filled syringe
2.
One pre-filled syringe contains 1,000 international units (IU) (8.3 µg) epoetin theta in 0.5 ml solution
for injection corresponding to 2,000 IU (16.7 µg) epoetin theta per ml.
Epoetin theta (recombinant human erythropoietin) is produced in Chinese Hamster Ovary Cells
(CHO-K1) by recombinant DNA technology.
For a full list of excipients, see section 6.1.
3.
Solution for injection (injection) in pre-filled syringe.
The solution is clear and colourless.
4.
-
Treatment of symptomatic anaemia associated with chronic renal failure in adult patients.
-
Treatment of symptomatic anaemia in adult cancer patients with non-myeloid malignancies
receiving chemotherapy.
Special requirements
Epoetin theta treatment should be initiated by physicians experienced in the above-mentioned
indications.
Routes of administration
The solution can be administered subcutaneously (SC) or intravenously (IV). Subcutaneous use is
preferable in patients who are not undergoing haemodialysis, in order to avoid puncturing peripheral
veins. If epoetin theta is substituted for another epoetin, the same route of administration should be
used. Epoetin theta should be administered by the subcutaneous route to cancer patients with
non-myeloid malignancies receiving chemotherapy.
Posology
Symptomatic anaemia associated
with chronic renal failure
Anaemia symptoms and sequelae may vary with age, gender, and overall burden of disease; a
physician’s evaluation of the individual patient’s clinical course and condition is necessary. Epoetin
theta should be administered either subcutaneously or intravenously in order to increase haemoglobin
level to not greater than 12 g/dl (7.45 mmol/l).
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.21 mmol/l) to 12 g/dl (7.45 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.45 mmol/l) should be avoided; guidance for appropriate dose adjustment if haemoglobin values
exceeding 12 g/dl (7.45 mmol/l) are observed are described below.
2
A rise in haemoglobin of greater than 2 g/dl (1.24 mmol/l) over a four week period should be avoided.
If the rise in haemoglobin is greater than 2 g/dl (1.24 mmol/l) in 4 weeks or the haemoglobin value
exceeds 12 g/dl (7.45 mmol/l), the dose should be reduced by 25 to 50%. It is recommended that
haemoglobin be monitored every two weeks until levels have stabilised and periodically thereafter. If
the haemoglobin level continues to increase, therapy should be interrupted until the haemoglobin level
begins to decrease, at which point therapy should be restarted at a dose approximately 25% below the
previously administered dose.
In the presence of hypertension or existing cardiovascular, cerebrovascular or peripheral vascular
diseases, the increase in haemoglobin and the target haemoglobin value should be determined
individually taking into account the clinical picture.
Treatment with epoetin theta is divided into two stages.
Correction phase
Subcutaneous administration: The initial posology is 20 IU/kg body weight 3 times per week. The
dose may be increased after 4 weeks to 40 IU/kg, 3 times per week, if the increase in haemoglobin is
not adequate (< 1 g/dl [0.62 mmol/l] within 4 weeks). Further increases of 25% of the previous dose
may be made at monthly intervals until the individual target haemoglobin level is obtained.
Intravenous administration: The initial posology is 40 IU/kg body weight 3 times per week. The dose
may be increased after 4 weeks to 80 IU/kg, 3 times per week, and by further increases of 25% of the
previous dose at monthly intervals, if needed.
For both routes of administration, the maximum dose should not exceed 700 IU/kg body weight per
week.
Maintenance phase
The dose should be adjusted as necessary to maintain the individual target haemoglobin level between
10 g/dl (6.21 mmol/l) to 12 g/dl (7.45 mmol/l), whereby a haemoglobin level of 12 g/dl (7.45 mmol/l)
should not be exceeded. If a dose adjustment is required to maintain the desired haemoglobin level, it
is recommended that the dose be adjusted by approximately 25%.
Subcutaneous administration: The weekly dose can be given as one injection per week or three times
per week.
Intravenous administration: Patients who are stable on a three times weekly dosing regimen may be
switched to twice-weekly administration.
If the frequency of administration is changed, haemoglobin level should be monitored closely and
dose adjustments may be necessary.
The maximum dose should not exceed 700 IU/kg body weight per week.
If epoetin theta is substituted for another epoetin, haemoglobin level should be monitored closely and
the same route of administration should be used.
Patients should be monitored closely to ensure that the lowest approved dose of epoetin theta is used
to provide adequate control of the symptoms of anaemia.
Symptomatic anaemia in cancer patients with non-myeloid malignancies receiving chemotherapy
Epoetin theta should be administered by the subcutaneous route to patients with anaemia (e.g.
haemoglobin concentration ≤ 10 g/dl [6.21 mmol/l]). Anaemia symptoms and sequelae may vary with
age, gender, and overall burden of disease; a physician’s evaluation of the individual patient’s clinical
course and condition is necessary.
3
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.21 mmol/l) to 12 g/dl (7.45 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.45 mmol/l) should be avoided; guidance for appropriate dose adjustment if haemoglobin values
exceeding 12 g/dl (7.45 mmol/l) are observed are described below.
The recommended initial dose is 20,000 IU, independent of bodyweight, given once-weekly. If, after
4 weeks of therapy, the haemoglobin value has increased by at least 1 g/dl (0.62 mmol/l), the current
dose should be continued. If the haemoglobin value has not increased by at least 1 g/dl (0.62 mmol/l) a
doubling of the weekly dose to 40,000 IU should be considered. If, after an additional 4 weeks of
therapy, the haemoglobin increase is still insufficient an increase of the weekly dose to 60,000 IU
should be considered.
The maximum dose should not exceed 60,000 IU per week.
If, after 12 weeks of therapy, the haemoglobin value has not increased by at least 1 g/dl (0.62 mmol/l),
response is unlikely and treatment should be discontinued.
If the rise in haemoglobin is greater than 2 g/dl (1.24 mmol/l) in 4 weeks or the haemoglobin level
exceeds 12 g/dl (7.45 mmol/l), the dose should be reduced by 25 to 50%. Treatment with epoetin theta
should be temporarily discontinued if haemoglobin levels exceed 13 g/dl (8.07 mmol/l). Therapy
should be reinitiated at approximately 25% lower than the previous dose after haemoglobin levels fall
to 12 g/dl (7.45 mmol/l) or below.
Therapy should be continued up to 4 weeks after the end of chemotherapy.
Patients should be monitored closely to ensure that the lowest approved dose of epoetin theta is used
to provide adequate control of the symptoms of anaemia.
Special populations
Paediatric patients
There is no experience in children and adolescents.
Method of administration
The solution can be administered subcutaneously or intravenously. Subcutaneous use is preferable in
patients who are not undergoing haemodialysis, in order to avoid puncturing peripheral veins. If
epoetin theta is substituted for another epoetin, the same route of administration should be used. In
cancer patients with non-myeloid malignancies receiving chemotherapy Epoetin theta should be
administered by the subcutaneous route only.
Subcutaneous injections should be given into the abdomen, arm or thigh.
Eporatio is supplied in a single use pre-filled syringe. The solution should be visually inspected prior
to use. Only clear, colourless solutions without particles should be used. The solution for injection
should not be shaken. It should be allowed to reach a comfortable temperature (15 °C - 25 °C) for
injection.
Eporatio must not be mixed with other medicinal products (see section 6.2).
The injection sites should be rotated and the injection performed slowly to avoid discomfort at the site
of injection.
-
Hypersensitivity to the active substance, other epoetins and derivatives or to any of the
excipients.
4
-
Uncontrolled hypertension.
General
Supplementary iron therapy is recommended for all patients with serum ferritin values below 100 µg/l
or with transferrin saturation below 20%. To ensure effective erythropoiesis, iron status has to be
evaluated for all patients prior to and during treatment.
Non-response to therapy with epoetin theta should prompt a search for causative factors. Deficiencies
of iron, folic acid or vitamin B
12
reduce the effectiveness of epoetins and should therefore be
corrected. Intercurrent infections, inflammatory or traumatic episodes, occult blood loss, haemolysis,
aluminium intoxication, underlying haematological diseases or bone marrow fibrosis may also
compromise the erythropoietic response. A reticulocyte count should be considered as part of the
evaluation.
Pure red cell aplasia (PRCA)
If typical causes of non-response are excluded, and the patient has a sudden drop in haemoglobin
associated with reticulocytopenia, an examination of anti-erythropoietin antibodies and the bone
marrow for diagnosis of pure red cell aplasia should be considered. Discontinuation of treatment with
epoetin theta should be taken into account.
PRCA caused by neutralising anti-erythropoietin antibodies has been reported in association with
erythropoietin therapy. These antibodies have been shown to cross-react with all epoetins, and patients
suspected or confirmed to have neutralising antibodies to erythropoietin should not be switched to
epoetin theta (see section 4.8).
A paradoxical decrease in haemoglobin and development of severe anaemia associated with low
reticulocyte counts should prompt to discontinue treatment with epoetin and perform
anti-erythropoietin antibody testing. Cases have been reported in patients with hepatitis C treated with
interferon and ribavirin, when epoetins are used concomitantly. Epoetins are not approved in the
management of anaemia associated with hepatitis C.
Hypertension
Patients on epoetin theta therapy can experience an increase in blood pressure or aggravation of
existing hypertension particularly during the initial treatment phase.
Therefore, in patients treated with epoetin theta, special care should be taken to monitor closely and
control blood pressure. Blood pressure should be controlled adequately before initiation and during
therapy to avoid acute complications, such as hypertensive crisis with encephalopathy-like symptoms
(e.g. headaches, confused state, speech disturbances, impaired gait) and related complications
(seizures, stroke), which may also occur in individual patients with otherwise normal or low blood
pressure. If these reactions occur, they require the immediate attention of a physician and intensive
medical care. Particular attention should be paid to sudden sharp migraine-like headaches as a possible
warning signal.
Increases in blood pressure may require treatment with antihypertensive medicinal products or a dose
increase of existing antihypertensive medicinal products. In addition, a reduction of the administered
dose of epoetin theta needs to be considered. If blood pressure values remain high, temporary
interruption of epoetin theta therapy may be required. Once hypertension has been controlled with
more intensified therapy, epoetin theta therapy should be re-started at a reduced dose.
Misuse
Misuse of epoetin theta by healthy persons may lead to an excessive increase in haemoglobin and
haematocrit. This may be associated with life-threatening cardiovascular complications.
5
Special populations
Due to limited experience, the efficacy and safety of epoetin theta could not be assessed in patients
with impaired liver function or homozygous sickle cell anaemia.
In clinical trials, patients over 75 years of age had a higher incidence of serious and severe adverse
events irrespective of a causal relationship to treatment with epoetin theta. Furthermore, deaths were
more frequent in this patient group compared to younger patients.
Laboratory monitoring
It is recommended that haemoglobin measurement, a complete blood count and platelet count be
performed regularly.
Symptomatic anaemia associated
with chronic renal failure
The use of epoetin theta in nephrosclerotic patients not yet undergoing dialysis should be defined
individually, as a possible accelerated progression of renal failure cannot be ruled out with certainty.
During haemodialysis, patients treated with epoetin theta may require increased anticoagulation
treatment to prevent clotting of the arterio-venous shunt.
In patients with chronic renal failure, the maintenance haemoglobin concentration should not exceed
the upper limit of the target haemoglobin concentration recommended in section 4.2. In clinical trials,
an increased risk of death and serious cardiovascular events was observed when epoetins were
administered to target a haemoglobin level in excess of 12 g/dl (7.45 mmol/l). Controlled clinical trials
have not shown significant benefits attributable to the administration of epoetins when the
haemoglobin concentration is increased beyond the level necessary to control symptoms of anaemia
and to avoid blood transfusion.
Symptomatic anaemia in cancer patients with non-myeloid malignancies receiving chemotherapy
Effect on tumour growth
Epoetins are growth factors that primarily stimulate red blood cell production. Erythropoietin
receptors may be expressed on the surface of a variety of tumour cells. As with all growth factors,
there is a concern that epoetins could stimulate the growth of any type of malignancy (see section 5.1).
In several controlled studies, epoetins have not been shown to improve overall survival or decrease the
risk of tumour progression in patients with anaemia associated with cancer. In controlled clinical
studies, use of epoetins has shown:
- shortened time to tumour progression in patients with advanced head and neck cancer receiving
radiation therapy when administered to target a haemoglobin level in excess of 14 g/dl
(8.69 mmol/l),
- shortened overall survival and increased deaths attributed to disease progression at 4 months in
patients with metastatic breast cancer receiving chemotherapy when administered to target a
haemoglobin value of 12-14 g/dl (7.45-8.69 mmol/l),
- increased risk of death when administered to target a haemoglobin value of 12 g/dl
(7.45 mmol/l) in patients with active malignant disease receiving neither chemotherapy nor
radiation therapy.
Epoetins are not indicated for use in this patient population.
In view of the above, in some clinical situations blood transfusion should be the preferred treatment
for the management of anaemia in patients with cancer. The decision to administer recombinant
erythropoietins should be based on a benefit-risk assessment with the participation of the individual
patient, which should take into account the specific clinical context. Factors that should be considered
in this assessment should include the type of tumour and its stage, the degree of anaemia,
life-expectancy, the environment in which the patient is being treated, and patient preference (see
section 5.1).
6
Excipients
This medicinal product contains less than 1 mmol sodium (23 mg) per pre-filled syringe, i.e.
essentially ‘sodium-free’.
No interaction studies have been performed.
For epoetin theta no clinical data on exposed pregnancies are available. Animal studies with other
epoetins do not indicate direct harmful effects with respect to pregnancy, embryonal/foetal
development, parturition or postnatal development (see section 5.3). Caution should be exercised
when prescribing to pregnant women.
It is unknown whether epoetin theta is excreted in human breast milk, but data in neonates show no
absorption or pharmacological activity of erythropoietin when given together with breast milk. A
decision on whether to continue/discontinue breast-feeding or to continue/discontinue therapy with
epoetin theta should be made taking into account the benefit of breast-feeding to the child and the
benefit of epoetin theta therapy for the woman.
Epoetin theta has no or negligible influence on the ability to drive and use machines.
The safety of epoetin theta has been evaluated based on results from clinical studies including
972 patients.
Approximately 9% of patients can be expected to experience an adverse reaction. The most frequent
undesirable effects are hypertension, influenza-like illness and headache.
Adverse reactions listed below are classified according to System Organ Class. Frequency groupings
are defined according to the following convention:
Very common: ≥ 1/10;
Common: ≥ 1/100 to < 1/10;
Uncommon: ≥ 1/1,000 to < 1/100;
Rare: ≥ 1/10,000 to < 1/1,000;
Very rare:
< 1/10,000;
Not known:
cannot be estimated from the available data.
7
System organ class
Adverse reaction
Frequency
Symptomatic anaemia
associated with
chronic renal failure
Symptomatic anaemia
in cancer patients with
non-myeloid
malignancies receiving
chemotherapy
Blood and lymphatic
system disorders
Thromboembolic
events
—
Not known
Shunt thrombosis
Common
—
Immune system
disorders
Hypersensitivity
reactions
Not known
Nervous system
disorders
Headache
Common
Vascular disorders
Hypertension
Common
Hypertensive crisis
Common
—
Skin and subcutaneous
tissue disorders
Skin reactions
Common
Musculoskeletal and
connective tissue
disorders
Arthralgia
—
Common
General disorders and
administration site
conditions
Influenza-like illness
Common
Shunt thrombosis may occur, especially in patients who have a tendency to hypotension or whose
arterio-venous fistulae exhibit complications (e.g. stenoses, aneurisms) (see section 4.4).
One of the most frequent adverse reactions during treatment with epoetin theta is an increase in blood
pressure or aggravation of existing hypertension particularly during the initial treatment phase.
Hypertension occurs in chronic renal failure patients more often during the correction phase than
during the maintenance phase. Hypertension can be treated with appropriate medicinal products (see
section 4.4).
Hypertensive crisis with encephalopathy-like symptoms (e.g. headaches, confused state, speech
disturbances, impaired gait) and related complications (seizures, stroke) may also occur in individual
patients with otherwise normal or low blood pressure (see section 4.4).
Skin reactions such as rash, pruritus or injection site reactions may occur.
Symptoms of influenza-like illness such as fever, chills and asthenic conditions have been reported.
Certain adverse reactions have not yet been observed with epoetin theta, but are generally accepted as
being attributable to epoetins:
In isolated cases in patients with chronic renal failure, neutralising anti-erythropoietin antibody-
mediated PRCA associated with therapy with other epoetins has been reported. If PRCA is diagnosed,
therapy with epoetin theta must be discontinued and patients should not be switched to another
recombinant epoetin (see section 4.4).
The therapeutic margin of epoetin theta is very wide. In the case of overdose, polycythaemia can
occur. In the event of polycythaemia, epoetin theta should be temporarily withheld.
If severe polycythaemia occurs, conventional methods (phlebotomy) may be indicated to reduce the
haemoglobin level.
8

















5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other anti-anaemic preparations, ATC code: B03XA01
Mechanism of action
Human erythropoietin is an endogenous glycoprotein hormone that is the primary regulator of
erythropoiesis through specific interaction with the erythropoietin receptor on the erythroid progenitor
cells in the bone marrow. It acts as a mitosis-stimulating factor and differentiation hormone. The
production of erythropoietin primarily occurs in and is regulated by the kidney in response to changes
in tissue oxygenation. Production of endogenous erythropoietin is impaired in patients with chronic
renal failure and the primary cause of their anaemia is erythropoietin deficiency. In patients with
cancer receiving chemotherapy the aetiology of anaemia is multifactorial. In these patients,
erythropoietin deficiency and a reduced response of erythroid progenitor cells to endogenous
erythropoietin both contribute significantly towards their anaemia.
Epoetin theta is identical in its amino acid sequence and similar in its carbohydrate composition
(glycosylation) to endogenous human erythropoietin.
Preclinical efficacy
The biological efficacy of epoetin theta has been demonstrated after intravenous and subcutaneous
administration in various animal models
in vivo
(mice, rats, dogs). After administration of epoetin
theta, the number of erythrocytes, the haematocrit values and reticulocyte counts increase.
Clinical efficacy and safety
Symptomatic anaemia associated chronic renal failure
Data from correction phase studies in 284 chronic renal failure patients show that the haemoglobin
response rates (defined as haemoglobin levels above 11 g/dl at two consecutive measurements) in the
epoetin theta group (88.4% and 89.4% in studies in patients on dialysis and not yet undergoing
dialysis, respectively) were comparable to epoetin beta (86.2% and 81.0%, respectively). The median
time to response was similar in the treatment groups with 56 days in haemodialysis patients and
49 days in patients not yet undergoing dialysis.
Two randomised controlled studies were conducted in 270 haemodialysis patients and 288 patients not
yet undergoing dialysis, who were on stable treatment with epoetin beta. Patients were randomised to
continue their current treatment or to be converted to epoetin theta (same dose as epoetin beta) in order
to maintain their haemoglobin levels. During the evaluation period (weeks 15 to 26), the mean and
median level of haemoglobin in patients treated with epoetin theta was virtually identical to their
baseline haemoglobin level. In these two studies, 180 haemodialysis patients and 193 patients not
undergoing dialysis were switched from maintenance phase treatment with epoetin beta to treatment
with epoetin theta for a period of six months showing stable haemoglobin values and a similar safety
profile as epoetin beta. In the clinical studies, patients not yet undergoing dialysis (subcutaneous
administration) discontinued the study more frequently than haemodialysis patients (intravenous
administration) as they had to terminate the study when starting dialysis.
In two long-term studies, the efficacy of epoetin theta was evaluated in 124 haemodialysis patients and
289 patients not yet undergoing dialysis. The haemoglobin levels remained within the desired target
range and epoetin theta was well tolerated over a period of up to 15 months.
In the clinical studies, pre-dialysis patients were treated once-weekly with epoetin theta, 174 patients
in the maintenance phase study and 111 patients in the long-term study.
Symptomatic anaemia in cancer patients with non-myeloid malignancies receiving chemotherapy
409 cancer patients receiving chemotherapy were included in two prospective, randomised
double-blind, placebo-controlled studies. The first study was conducted in 186 anaemic patients with
9
non-myeloid malignancies (55% with haematological malignancies and 45% with solid tumours)
receiving non-platinum chemotherapy. The second study was conducted in 223 patients with various
solid tumours receiving platinum-containing chemotherapy. In both studies, treatment with epoetin
theta resulted in a significant haemoglobin response (p < 0.001), defined as an increase in
haemoglobin of ≥ 2 g/dl without transfusion, and a significant reduction in transfusion requirements
(p < 0.05) in comparison to placebo.
Effect on tumour growth
Erythropoietin is a growth factor that primarily stimulates red cell production. Erythropoietin receptors
may be expressed on the surface of a variety of tumour cells.
Survival and tumour progression have been examined in five large controlled studies involving a total
of 2,833 patients, of which four were double-blind placebo-controlled studies and one was an open-
label study. Two of the studies recruited patients who were being treated with chemotherapy. The
target haemoglobin concentration in two studies was > 13 g/dl; in the remaining three studies it was
12-14 g/dl. In the open-label study there was no difference in overall survival between patients treated
with recombinant human erythropoietin and controls. In the four placebo-controlled studies the hazard
ratios for overall survival ranged between 1.25 and 2.47 in favour of controls. These studies have
shown a consistent unexplained statistically significant excess mortality in patients who have anaemia
associated with various common cancers who received recombinant human erythropoietin compared
to controls. Overall survival outcome in the trials could not be satisfactorily explained by differences
in the incidence of thrombosis and related complications between those given recombinant human
erythropoietin and those in the control group.
Data from three placebo-controlled clinical studies in 586 anaemic cancer patients conducted with
epoetin theta, showed no negative effect of epoetin theta on survival. During the studies, mortality was
lower in the epoetin theta group (6.9%) compared to placebo (10.3%).
A systematic review has also been performed involving more than 9,000 cancer patients participating
in 57 clinical trials. Meta-analysis of overall survival data produced a hazard ratio point estimate of
1.08 in favour of controls (95% CI: 0.99, 1.18; 42 trials and 8,167 patients). An increased relative risk
of thromboembolic events (RR 1.67, 95% CI: 1.35, 2.06; 35 trials and 6,769 patients) was observed in
patients treated with recombinant human erythropoietin. There is therefore consistent evidence to
suggest that there may be significant harm to patients with cancer who are treated with recombinant
human erythropoietin. The extent to which these outcomes might apply to the administration of
recombinant human erythropoietin to patients with cancer, treated with chemotherapy to achieve
haemoglobin concentrations less than 13 g/dl, is unclear because few patients with these
characteristics were included in the data reviewed.
A patient-level data analysis has also been performed on more than 13,900 cancer patients (chemo-,
radio-, chemoradio- or no therapy) participating in 53 controlled clinical trials involving several
epoetins. Meta-analysis of overall survival data produced a hazard ratio point estimate of 1.06 in
favour of controls (95% CI: 1.00, 1.12; 53 trials and 13,933 patients) and for cancer patients receiving
chemotherapy, the overall survival hazard ratio was 1.04 (95% CI: 0.97, 1.11; 38 trials and 10,441
patients). Meta-analyses also indicate consistently a significantly increased relative risk of
thromboembolic events in cancer patients receiving recombinant human erythropoietin (see section
4.4).
5.2 Pharmacokinetic properties
General
The pharmacokinetics of epoetin theta have been examined in healthy volunteers, in patients with
chronic renal failure and in cancer patients receiving chemotherapy. The pharmacokinetics of epoetin
theta are independent of age or gender.
Subcutaneous administration
10
Following subcutaneous injection of 40 IU/kg body weight epoetin theta at three different sites (upper
arm, abdomen, thigh) in healthy volunteers, similar plasma level profiles were observed. The extent of
absorption (AUC) was slightly greater after injection in the abdomen in comparison to the other sites.
The maximum concentration is reached after an average of 10 to 14 hours and the average terminal
half-life ranges from approximately 22 to 41 hours.
Average bioavailability of epoetin theta after subcutaneous administration is approximately 31%
compared with intravenous administration.
In pre-dialysis patients with chronic renal failure following subcutaneous injection of 40 IU/kg body
weight, the protracted absorption results in a concentration plateau, whereby the maximum
concentration is reached after an average of approximately 14 hours. The terminal half-life is higher
than after intravenous administration, with an average of 25 hours after single dosing and 34 hours in
steady state after repeated dosing three times weekly, without leading to an accumulation of epoetin
theta.
In cancer patients receiving chemotherapy, after repeated subcutaneous administration of 20,000 IU
epoetin theta once-weekly, the terminal half-life is 29 hours after the first dose and 28 hours in steady
state. No accumulation of epoetin theta was observed.
Intravenous administration
In patients with chronic renal failure undergoing haemodialysis, the elimination half-life of epoetin
theta is 6 hours after single dosing and 4 hours in steady state after repeated intravenous administration
of 40 IU/kg body weight epoetin theta three times weekly. No accumulation of epoetin theta was
observed. Following intravenous administration, the volume of distribution approximates to total
blood volume.
5.3 Preclinical safety data
Non-clinical data with epoetin theta reveal no special hazard for humans based on conventional studies
of safety pharmacology and repeated-dose toxicity.
Non-clinical data with other epoetins reveal no special hazard for humans based on conventional
studies of genotoxicity and toxicity to reproduction.
In reproductive toxicity studies performed with other epoetins, effects interpreted as being secondary
to decreased maternal body weight were observed at doses sufficiently in excess to the recommended
human dose.
6.
6.1 List of excipients
Sodium dihydrogen phosphate dihydrate
Sodium chloride
Polysorbate 20
Trometamol
Hydrochloric acid (6 M) (for pH adjustment)
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3 Shelf life
11
2 years.
6.4 Special precautions for storage
Store in a refrigerator (2 °C – 8 °C).
Do not freeze.
Keep the pre-filled syringe in the outer carton in order to protect from light.
For the purpose of ambulatory use, the patient may remove the product from the refrigerator and store
it at a temperature not above 25 °C for a single period of up to 7 days without exceeding the expiry
date. Once removed from the refrigerator, the medicinal product must be used within this period or
disposed of.
6.5
Nature and contents of container
0.5 ml of solution in a pre-filled syringe (type I glass) with a tip cap (bromobutyl rubber), a plunger
stopper (teflonised chlorobutyl rubber), an injection needle (stainless steel) and with or without a
pre-assembled safety device.
Pack sizes of 6 pre-filled syringes with or without safety device.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
The pre-filled syringes are for single use only.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
ratiopharm GmbH
Graf-Arco-Straße 3
89079 Ulm
Germany
info@ratiopharm.de
8.
EU/1/09/573/001
EU/1/09/573/002
9.
Date of first authorisation: 29 October 2009.
12
Detailed information on this medicinal product is available on the website of the European Medicines
Agency (EMEA)
http://www.emea.europa.eu/
.
13
1.
Eporatio 2,000 IU/0.5 ml solution for injection in pre-filled syringe
2.
One pre-filled syringe contains 2,000 international units (IU) (16.7 µg) epoetin theta in 0.5 ml solution
for injection corresponding to 4,000 IU (33.3 µg) epoetin theta per ml.
Epoetin theta (recombinant human erythropoietin) is produced in Chinese Hamster Ovary Cells
(CHO-K1) by recombinant DNA technology.
For a full list of excipients, see section 6.1.
3.
Solution for injection (injection) in pre-filled syringe.
The solution is clear and colourless.
4.
-
Treatment of symptomatic anaemia associated with chronic renal failure in adult patients.
-
Treatment of symptomatic anaemia in adult cancer patients with non-myeloid malignancies
receiving chemotherapy.
Special requirements
Epoetin theta treatment should be initiated by physicians experienced in the above-mentioned
indications.
Routes of administration
The solution can be administered subcutaneously (SC) or intravenously (IV). Subcutaneous use is
preferable in patients who are not undergoing haemodialysis, in order to avoid puncturing peripheral
veins. If epoetin theta is substituted for another epoetin, the same route of administration should be
used. Epoetin theta should be administered by the subcutaneous route to cancer patients with
non-myeloid malignancies receiving chemotherapy.
Posology
Symptomatic anaemia associated
with chronic renal failure
Anaemia symptoms and sequelae may vary with age, gender, and overall burden of disease; a
physician’s evaluation of the individual patient’s clinical course and condition is necessary. Epoetin
theta should be administered either subcutaneously or intravenously in order to increase haemoglobin
level to not greater than 12 g/dl (7.45 mmol/l).
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.21 mmol/l) to 12 g/dl (7.45 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.45 mmol/l) should be avoided; guidance for appropriate dose adjustment if haemoglobin values
exceeding 12 g/dl (7.45 mmol/l) are observed are described below.
14
A rise in haemoglobin of greater than 2 g/dl (1.24 mmol/l) over a four week period should be avoided.
If the rise in haemoglobin is greater than 2 g/dl (1.24 mmol/l) in 4 weeks or the haemoglobin value
exceeds 12 g/dl (7.45 mmol/l), the dose should be reduced by 25 to 50%. It is recommended that
haemoglobin be monitored every two weeks until levels have stabilised and periodically thereafter. If
the haemoglobin level continues to increase, therapy should be interrupted until the haemoglobin level
begins to decrease, at which point therapy should be restarted at a dose approximately 25% below the
previously administered dose.
In the presence of hypertension or existing cardiovascular, cerebrovascular or peripheral vascular
diseases, the increase in haemoglobin and the target haemoglobin value should be determined
individually taking into account the clinical picture.
Treatment with epoetin theta is divided into two stages.
Correction phase
Subcutaneous administration: The initial posology is 20 IU/kg body weight 3 times per week. The
dose may be increased after 4 weeks to 40 IU/kg, 3 times per week, if the increase in haemoglobin is
not adequate (< 1 g/dl [0.62 mmol/l] within 4 weeks). Further increases of 25% of the previous dose
may be made at monthly intervals until the individual target haemoglobin level is obtained.
Intravenous administration: The initial posology is 40 IU/kg body weight 3 times per week. The dose
may be increased after 4 weeks to 80 IU/kg, 3 times per week, and by further increases of 25% of the
previous dose at monthly intervals, if needed.
For both routes of administration, the maximum dose should not exceed 700 IU/kg body weight per
week.
Maintenance phase
The dose should be adjusted as necessary to maintain the individual target haemoglobin level between
10 g/dl (6.21 mmol/l) to 12 g/dl (7.45 mmol/l), whereby a haemoglobin level of 12 g/dl (7.45 mmol/l)
should not be exceeded. If a dose adjustment is required to maintain the desired haemoglobin level, it
is recommended that the dose be adjusted by approximately 25%.
Subcutaneous administration: The weekly dose can be given as one injection per week or three times
per week.
Intravenous administration: Patients who are stable on a three times weekly dosing regimen may be
switched to twice-weekly administration.
If the frequency of administration is changed, haemoglobin level should be monitored closely and
dose adjustments may be necessary.
The maximum dose should not exceed 700 IU/kg body weight per week.
If epoetin theta is substituted for another epoetin, haemoglobin level should be monitored closely and
the same route of administration should be used.
Patients should be monitored closely to ensure that the lowest approved dose of epoetin theta is used
to provide adequate control of the symptoms of anaemia.
Symptomatic anaemia in cancer patients with non-myeloid malignancies receiving chemotherapy
Epoetin theta should be administered by the subcutaneous route to patients with anaemia (e.g.
haemoglobin concentration ≤ 10 g/dl [6.21 mmol/l]). Anaemia symptoms and sequelae may vary with
age, gender, and overall burden of disease; a physician’s evaluation of the individual patient’s clinical
course and condition is necessary.
15
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.21 mmol/l) to 12 g/dl (7.45 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.45 mmol/l) should be avoided; guidance for appropriate dose adjustment if haemoglobin values
exceeding 12 g/dl (7.45 mmol/l) are observed are described below.
The recommended initial dose is 20,000 IU, independent of bodyweight, given once-weekly. If, after
4 weeks of therapy, the haemoglobin value has increased by at least 1 g/dl (0.62 mmol/l), the current
dose should be continued. If the haemoglobin value has not increased by at least 1 g/dl (0.62 mmol/l) a
doubling of the weekly dose to 40,000 IU should be considered. If, after an additional 4 weeks of
therapy, the haemoglobin increase is still insufficient an increase of the weekly dose to 60,000 IU
should be considered.
The maximum dose should not exceed 60,000 IU per week.
If, after 12 weeks of therapy, the haemoglobin value has not increased by at least 1 g/dl (0.62 mmol/l),
response is unlikely and treatment should be discontinued.
If the rise in haemoglobin is greater than 2 g/dl (1.24 mmol/l) in 4 weeks or the haemoglobin level
exceeds 12 g/dl (7.45 mmol/l), the dose should be reduced by 25 to 50%. Treatment with epoetin theta
should be temporarily discontinued if haemoglobin levels exceed 13 g/dl (8.07 mmol/l). Therapy
should be reinitiated at approximately 25% lower than the previous dose after haemoglobin levels fall
to 12 g/dl (7.45 mmol/l) or below.
Therapy should be continued up to 4 weeks after the end of chemotherapy.
Patients should be monitored closely to ensure that the lowest approved dose of epoetin theta is used
to provide adequate control of the symptoms of anaemia.
Special populations
Paediatric patients
There is no experience in children and adolescents.
Method of administration
The solution can be administered subcutaneously or intravenously. Subcutaneous use is preferable in
patients who are not undergoing haemodialysis, in order to avoid puncturing peripheral veins. If
epoetin theta is substituted for another epoetin, the same route of administration should be used. In
cancer patients with non-myeloid malignancies receiving chemotherapy Epoetin theta should be
administered by the subcutaneous route only.
Subcutaneous injections should be given into the abdomen, arm or thigh.
Eporatio is supplied in a single use pre-filled syringe. The solution should be visually inspected prior
to use. Only clear, colourless solutions without particles should be used. The solution for injection
should not be shaken. It should be allowed to reach a comfortable temperature (15 °C - 25 °C) for
injection.
Eporatio must not be mixed with other medicinal products (see section 6.2).
The injection sites should be rotated and the injection performed slowly to avoid discomfort at the site
of injection.
-
Hypersensitivity to the active substance, other epoetins and derivatives or to any of the
excipients.
16
-
Uncontrolled hypertension.
General
Supplementary iron therapy is recommended for all patients with serum ferritin values below 100 µg/l
or with transferrin saturation below 20%. To ensure effective erythropoiesis, iron status has to be
evaluated for all patients prior to and during treatment.
Non-response to therapy with epoetin theta should prompt a search for causative factors. Deficiencies
of iron, folic acid or vitamin B
12
reduce the effectiveness of epoetins and should therefore be
corrected. Intercurrent infections, inflammatory or traumatic episodes, occult blood loss, haemolysis,
aluminium intoxication, underlying haematological diseases or bone marrow fibrosis may also
compromise the erythropoietic response. A reticulocyte count should be considered as part of the
evaluation.
Pure red cell aplasia (PRCA)
If typical causes of non-response are excluded, and the patient has a sudden drop in haemoglobin
associated with reticulocytopenia, an examination of anti-erythropoietin antibodies and the bone
marrow for diagnosis of pure red cell aplasia should be considered. Discontinuation of treatment with
epoetin theta should be taken into account.
PRCA caused by neutralising anti-erythropoietin antibodies has been reported in association with
erythropoietin therapy. These antibodies have been shown to cross-react with all epoetins, and patients
suspected or confirmed to have neutralising antibodies to erythropoietin should not be switched to
epoetin theta (see section 4.8).
A paradoxical decrease in haemoglobin and development of severe anaemia associated with low
reticulocyte counts should prompt to discontinue treatment with epoetin and perform
anti-erythropoietin antibody testing. Cases have been reported in patients with hepatitis C treated with
interferon and ribavirin, when epoetins are used concomitantly. Epoetins are not approved in the
management of anaemia associated with hepatitis C.
Hypertension
Patients on epoetin theta therapy can experience an increase in blood pressure or aggravation of
existing hypertension particularly during the initial treatment phase.
Therefore, in patients treated with epoetin theta, special care should be taken to monitor closely and
control blood pressure. Blood pressure should be controlled adequately before initiation and during
therapy to avoid acute complications, such as hypertensive crisis with encephalopathy-like symptoms
(e.g. headaches, confused state, speech disturbances, impaired gait) and related complications
(seizures, stroke), which may also occur in individual patients with otherwise normal or low blood
pressure. If these reactions occur, they require the immediate attention of a physician and intensive
medical care. Particular attention should be paid to sudden sharp migraine-like headaches as a possible
warning signal.
Increases in blood pressure may require treatment with antihypertensive medicinal products or a dose
increase of existing antihypertensive medicinal products. In addition, a reduction of the administered
dose of epoetin theta needs to be considered. If blood pressure values remain high, temporary
interruption of epoetin theta therapy may be required. Once hypertension has been controlled with
more intensified therapy, epoetin theta therapy should be re-started at a reduced dose.
Misuse
Misuse of epoetin theta by healthy persons may lead to an excessive increase in haemoglobin and
haematocrit. This may be associated with life-threatening cardiovascular complications.
17
Special populations
Due to limited experience, the efficacy and safety of epoetin theta could not be assessed in patients
with impaired liver function or homozygous sickle cell anaemia.
In clinical trials, patients over 75 years of age had a higher incidence of serious and severe adverse
events irrespective of a causal relationship to treatment with epoetin theta. Furthermore, deaths were
more frequent in this patient group compared to younger patients.
Laboratory monitoring
It is recommended that haemoglobin measurement, a complete blood count and platelet count be
performed regularly.
Symptomatic anaemia associated
with chronic renal failure
The use of epoetin theta in nephrosclerotic patients not yet undergoing dialysis should be defined
individually, as a possible accelerated progression of renal failure cannot be ruled out with certainty.
During haemodialysis, patients treated with epoetin theta may require increased anticoagulation
treatment to prevent clotting of the arterio-venous shunt.
In patients with chronic renal failure, the maintenance haemoglobin concentration should not exceed
the upper limit of the target haemoglobin concentration recommended in section 4.2. In clinical trials,
an increased risk of death and serious cardiovascular events was observed when epoetins were
administered to target a haemoglobin level in excess of 12 g/dl (7.45 mmol/l). Controlled clinical trials
have not shown significant benefits attributable to the administration of epoetins when the
haemoglobin concentration is increased beyond the level necessary to control symptoms of anaemia
and to avoid blood transfusion.
Symptomatic anaemia in cancer patients with non-myeloid malignancies receiving chemotherapy
Effect on tumour growth
Epoetins are growth factors that primarily stimulate red blood cell production. Erythropoietin
receptors may be expressed on the surface of a variety of tumour cells. As with all growth factors,
there is a concern that epoetins could stimulate the growth of any type of malignancy (see section 5.1).
In several controlled studies, epoetins have not been shown to improve overall survival or decrease the
risk of tumour progression in patients with anaemia associated with cancer. In controlled clinical
studies, use of epoetins has shown:
- shortened time to tumour progression in patients with advanced head and neck cancer receiving
radiation therapy when administered to target a haemoglobin level in excess of 14 g/dl
(8.69 mmol/l),
- shortened overall survival and increased deaths attributed to disease progression at 4 months in
patients with metastatic breast cancer receiving chemotherapy when administered to target a
haemoglobin value of 12-14 g/dl (7.45-8.69 mmol/l),
- increased risk of death when administered to target a haemoglobin value of 12 g/dl
(7.45 mmol/l) in patients with active malignant disease receiving neither chemotherapy nor
radiation therapy.
Epoetins are not indicated for use in this patient population.
In view of the above, in some clinical situations blood transfusion should be the preferred treatment
for the management of anaemia in patients with cancer. The decision to administer recombinant
erythropoietins should be based on a benefit-risk assessment with the participation of the individual
patient, which should take into account the specific clinical context. Factors that should be considered
in this assessment should include the type of tumour and its stage, the degree of anaemia,
life-expectancy, the environment in which the patient is being treated, and patient preference (see
section 5.1).
18
Excipients
This medicinal product contains less than 1 mmol sodium (23 mg) per pre-filled syringe, i.e.
essentially ‘sodium-free’.
No interaction studies have been performed.
For epoetin theta no clinical data on exposed pregnancies are available. Animal studies with other
epoetins do not indicate direct harmful effects with respect to pregnancy, embryonal/foetal
development, parturition or postnatal development (see section 5.3). Caution should be exercised
when prescribing to pregnant women.
It is unknown whether epoetin theta is excreted in human breast milk, but data in neonates show no
absorption or pharmacological activity of erythropoietin when given together with breast milk. A
decision on whether to continue/discontinue breast-feeding or to continue/discontinue therapy with
epoetin theta should be made taking into account the benefit of breast-feeding to the child and the
benefit of epoetin theta therapy for the woman.
Epoetin theta has no or negligible influence on the ability to drive and use machines.
The safety of epoetin theta has been evaluated based on results from clinical studies including
972 patients.
Approximately 9% of patients can be expected to experience an adverse reaction. The most frequent
undesirable effects are hypertension, influenza-like illness and headache.
Adverse reactions listed below are classified according to System Organ Class. Frequency groupings
are defined according to the following convention:
Very common: ≥ 1/10;
Common: ≥ 1/100 to < 1/10;
Uncommon: ≥ 1/1,000 to < 1/100;
Rare: ≥ 1/10,000 to < 1/1,000;
Very rare:
< 1/10,000;
Not known:
cannot be estimated from the available data.
19
System organ class
Adverse reaction
Frequency
Symptomatic anaemia
associated with
chronic renal failure
Symptomatic anaemia
in cancer patients with
non-myeloid
malignancies receiving
chemotherapy
Blood and lymphatic
system disorders
Thromboembolic
events
—
Not known
Shunt thrombosis
Common
—
Immune system
disorders
Hypersensitivity
reactions
Not known
Nervous system
disorders
Headache
Common
Vascular disorders
Hypertension
Common
Hypertensive crisis
Common
—
Skin and subcutaneous
tissue disorders
Skin reactions
Common
Musculoskeletal and
connective tissue
disorders
Arthralgia
—
Common
General disorders and
administration site
conditions
Influenza-like illness
Common
Shunt thrombosis may occur, especially in patients who have a tendency to hypotension or whose
arterio-venous fistulae exhibit complications (e.g. stenoses, aneurisms) (see section 4.4).
One of the most frequent adverse reactions during treatment with epoetin theta is an increase in blood
pressure or aggravation of existing hypertension particularly during the initial treatment phase.
Hypertension occurs in chronic renal failure patients more often during the correction phase than
during the maintenance phase. Hypertension can be treated with appropriate medicinal products (see
section 4.4).
Hypertensive crisis with encephalopathy-like symptoms (e.g. headaches, confused state, speech
disturbances, impaired gait) and related complications (seizures, stroke) may also occur in individual
patients with otherwise normal or low blood pressure (see section 4.4).
Skin reactions such as rash, pruritus or injection site reactions may occur.
Symptoms of influenza-like illness such as fever, chills and asthenic conditions have been reported.
Certain adverse reactions have not yet been observed with epoetin theta, but are generally accepted as
being attributable to epoetins:
In isolated cases in patients with chronic renal failure, neutralising anti-erythropoietin antibody-
mediated PRCA associated with therapy with other epoetins has been reported. If PRCA is diagnosed,
therapy with epoetin theta must be discontinued and patients should not be switched to another
recombinant epoetin (see section 4.4).
The therapeutic margin of epoetin theta is very wide. In the case of overdose, polycythaemia can
occur. In the event of polycythaemia, epoetin theta should be temporarily withheld.
If severe polycythaemia occurs, conventional methods (phlebotomy) may be indicated to reduce the
haemoglobin level.
20

















5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other anti-anaemic preparations, ATC code: B03XA01
Mechanism of action
Human erythropoietin is an endogenous glycoprotein hormone that is the primary regulator of
erythropoiesis through specific interaction with the erythropoietin receptor on the erythroid progenitor
cells in the bone marrow. It acts as a mitosis-stimulating factor and differentiation hormone. The
production of erythropoietin primarily occurs in and is regulated by the kidney in response to changes
in tissue oxygenation. Production of endogenous erythropoietin is impaired in patients with chronic
renal failure and the primary cause of their anaemia is erythropoietin deficiency. In patients with
cancer receiving chemotherapy the aetiology of anaemia is multifactorial. In these patients,
erythropoietin deficiency and a reduced response of erythroid progenitor cells to endogenous
erythropoietin both contribute significantly towards their anaemia.
Epoetin theta is identical in its amino acid sequence and similar in its carbohydrate composition
(glycosylation) to endogenous human erythropoietin.
Preclinical efficacy
The biological efficacy of epoetin theta has been demonstrated after intravenous and subcutaneous
administration in various animal models
in vivo
(mice, rats, dogs). After administration of epoetin
theta, the number of erythrocytes, the haematocrit values and reticulocyte counts increase.
Clinical efficacy and safety
Symptomatic anaemia associated chronic renal failure
Data from correction phase studies in 284 chronic renal failure patients show that the haemoglobin
response rates (defined as haemoglobin levels above 11 g/dl at two consecutive measurements) in the
epoetin theta group (88.4% and 89.4% in studies in patients on dialysis and not yet undergoing
dialysis, respectively) were comparable to epoetin beta (86.2% and 81.0%, respectively). The median
time to response was similar in the treatment groups with 56 days in haemodialysis patients and
49 days in patients not yet undergoing dialysis.
Two randomised controlled studies were conducted in 270 haemodialysis patients and 288 patients not
yet undergoing dialysis, who were on stable treatment with epoetin beta. Patients were randomised to
continue their current treatment or to be converted to epoetin theta (same dose as epoetin beta) in order
to maintain their haemoglobin levels. During the evaluation period (weeks 15 to 26), the mean and
median level of haemoglobin in patients treated with epoetin theta was virtually identical to their
baseline haemoglobin level. In these two studies, 180 haemodialysis patients and 193 patients not
undergoing dialysis were switched from maintenance phase treatment with epoetin beta to treatment
with epoetin theta for a period of six months showing stable haemoglobin values and a similar safety
profile as epoetin beta. In the clinical studies, patients not yet undergoing dialysis (subcutaneous
administration) discontinued the study more frequently than haemodialysis patients (intravenous
administration) as they had to terminate the study when starting dialysis.
In two long-term studies, the efficacy of epoetin theta was evaluated in 124 haemodialysis patients and
289 patients not yet undergoing dialysis. The haemoglobin levels remained within the desired target
range and epoetin theta was well tolerated over a period of up to 15 months.
In the clinical studies, pre-dialysis patients were treated once-weekly with epoetin theta, 174 patients
in the maintenance phase study and 111 patients in the long-term study.
Symptomatic anaemia in cancer patients with non-myeloid malignancies receiving chemotherapy
409 cancer patients receiving chemotherapy were included in two prospective, randomised
double-blind, placebo-controlled studies. The first study was conducted in 186 anaemic patients with
21
non-myeloid malignancies (55% with haematological malignancies and 45% with solid tumours)
receiving non-platinum chemotherapy. The second study was conducted in 223 patients with various
solid tumours receiving platinum-containing chemotherapy. In both studies, treatment with epoetin
theta resulted in a significant haemoglobin response (p < 0.001), defined as an increase in
haemoglobin of ≥ 2 g/dl without transfusion, and a significant reduction in transfusion requirements
(p < 0.05) in comparison to placebo.
Effect on tumour growth
Erythropoietin is a growth factor that primarily stimulates red cell production. Erythropoietin receptors
may be expressed on the surface of a variety of tumour cells.
Survival and tumour progression have been examined in five large controlled studies involving a total
of 2,833 patients, of which four were double-blind placebo-controlled studies and one was an open-
label study. Two of the studies recruited patients who were being treated with chemotherapy. The
target haemoglobin concentration in two studies was > 13 g/dl; in the remaining three studies it was
12-14 g/dl. In the open-label study there was no difference in overall survival between patients treated
with recombinant human erythropoietin and controls. In the four placebo-controlled studies the hazard
ratios for overall survival ranged between 1.25 and 2.47 in favour of controls. These studies have
shown a consistent unexplained statistically significant excess mortality in patients who have anaemia
associated with various common cancers who received recombinant human erythropoietin compared
to controls. Overall survival outcome in the trials could not be satisfactorily explained by differences
in the incidence of thrombosis and related complications between those given recombinant human
erythropoietin and those in the control group.
Data from three placebo-controlled clinical studies in 586 anaemic cancer patients conducted with
epoetin theta, showed no negative effect of epoetin theta on survival. During the studies, mortality was
lower in the epoetin theta group (6.9%) compared to placebo (10.3%).
A systematic review has also been performed involving more than 9,000 cancer patients participating
in 57 clinical trials. Meta-analysis of overall survival data produced a hazard ratio point estimate of
1.08 in favour of controls (95% CI: 0.99, 1.18; 42 trials and 8,167 patients). An increased relative risk
of thromboembolic events (RR 1.67, 95% CI: 1.35, 2.06; 35 trials and 6,769 patients) was observed in
patients treated with recombinant human erythropoietin. There is therefore consistent evidence to
suggest that there may be significant harm to patients with cancer who are treated with recombinant
human erythropoietin. The extent to which these outcomes might apply to the administration of
recombinant human erythropoietin to patients with cancer, treated with chemotherapy to achieve
haemoglobin concentrations less than 13 g/dl, is unclear because few patients with these
characteristics were included in the data reviewed.
A patient-level data analysis has also been performed on more than 13,900 cancer patients (chemo-,
radio-, chemoradio- or no therapy) participating in 53 controlled clinical trials involving several
epoetins. Meta-analysis of overall survival data produced a hazard ratio point estimate of 1.06 in
favour of controls (95% CI: 1.00, 1.12; 53 trials and 13,933 patients) and for cancer patients receiving
chemotherapy, the overall survival hazard ratio was 1.04 (95% CI: 0.97, 1.11; 38 trials and 10,441
patients). Meta-analyses also indicate consistently a significantly increased relative risk of
thromboembolic events in cancer patients receiving recombinant human erythropoietin (see section
4.4).
5.2 Pharmacokinetic properties
General
The pharmacokinetics of epoetin theta have been examined in healthy volunteers, in patients with
chronic renal failure and in cancer patients receiving chemotherapy. The pharmacokinetics of epoetin
theta are independent of age or gender.
Subcutaneous administration
22
Following subcutaneous injection of 40 IU/kg body weight epoetin theta at three different sites (upper
arm, abdomen, thigh) in healthy volunteers, similar plasma level profiles were observed. The extent of
absorption (AUC) was slightly greater after injection in the abdomen in comparison to the other sites.
The maximum concentration is reached after an average of 10 to 14 hours and the average terminal
half-life ranges from approximately 22 to 41 hours.
Average bioavailability of epoetin theta after subcutaneous administration is approximately 31%
compared with intravenous administration.
In pre-dialysis patients with chronic renal failure following subcutaneous injection of 40 IU/kg body
weight, the protracted absorption results in a concentration plateau, whereby the maximum
concentration is reached after an average of approximately 14 hours. The terminal half-life is higher
than after intravenous administration, with an average of 25 hours after single dosing and 34 hours in
steady state after repeated dosing three times weekly, without leading to an accumulation of epoetin
theta.
In cancer patients receiving chemotherapy, after repeated subcutaneous administration of 20,000 IU
epoetin theta once-weekly, the terminal half-life is 29 hours after the first dose and 28 hours in steady
state. No accumulation of epoetin theta was observed.
Intravenous administration
In patients with chronic renal failure undergoing haemodialysis, the elimination half-life of epoetin
theta is 6 hours after single dosing and 4 hours in steady state after repeated intravenous administration
of 40 IU/kg body weight epoetin theta three times weekly. No accumulation of epoetin theta was
observed. Following intravenous administration, the volume of distribution approximates to total
blood volume.
5.3 Preclinical safety data
Non-clinical data with epoetin theta reveal no special hazard for humans based on conventional studies
of safety pharmacology and repeated-dose toxicity.
Non-clinical data with other epoetins reveal no special hazard for humans based on conventional
studies of genotoxicity and toxicity to reproduction.
In reproductive toxicity studies performed with other epoetins, effects interpreted as being secondary
to decreased maternal body weight were observed at doses sufficiently in excess to the recommended
human dose.
6.
6.1 List of excipients
Sodium dihydrogen phosphate dihydrate
Sodium chloride
Polysorbate 20
Trometamol
Hydrochloric acid (6 M) (for pH adjustment)
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3 Shelf life
23
2 years.
6.4 Special precautions for storage
Store in a refrigerator (2 °C – 8 °C).
Do not freeze.
Keep the pre-filled syringe in the outer carton in order to protect from light.
For the purpose of ambulatory use, the patient may remove the product from the refrigerator and store
it at a temperature not above 25 °C for a single period of up to 7 days without exceeding the expiry
date. Once removed from the refrigerator, the medicinal product must be used within this period or
disposed of.
6.5 Nature and contents of container
0.5 ml of solution in a pre-filled syringe (type I glass) with a tip cap (bromobutyl rubber), a plunger
stopper (teflonised chlorobutyl rubber), an injection needle (stainless steel) and with or without a
pre-assembled safety device.
Pack sizes of 6 pre-filled syringes with or without safety device.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
The pre-filled syringes are for single use only.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
ratiopharm GmbH
Graf-Arco-Straße 3
89079 Ulm
Germany
info@ratiopharm.de
8.
EU/1/09/573/003
EU/1/09/573/004
9.
Date of first authorisation: 29 October 2009.
24
Detailed information on this medicinal product is available on the website of the European Medicines
Agency (EMEA)
http://www.emea.europa.eu/
.
25
1.
Eporatio 3,000 IU/0.5 ml solution for injection in pre-filled syringe
2.
One pre-filled syringe contains 3,000 international units (IU) (25 µg) epoetin theta in 0.5 ml solution
for injection corresponding to 6,000 IU (50 µg) epoetin theta per ml.
Epoetin theta (recombinant human erythropoietin) is produced in Chinese Hamster Ovary Cells
(CHO-K1) by recombinant DNA technology.
For a full list of excipients, see section 6.1.
3.
Solution for injection (injection) in pre-filled syringe.
The solution is clear and colourless.
4.
-
Treatment of symptomatic anaemia associated with chronic renal failure in adult patients.
-
Treatment of symptomatic anaemia in adult cancer patients with non-myeloid malignancies
receiving chemotherapy.
Special requirements
Epoetin theta treatment should be initiated by physicians experienced in the above-mentioned
indications.
Routes of administration
The solution can be administered subcutaneously (SC) or intravenously (IV). Subcutaneous use is
preferable in patients who are not undergoing haemodialysis, in order to avoid puncturing peripheral
veins. If epoetin theta is substituted for another epoetin, the same route of administration should be
used. Epoetin theta should be administered by the subcutaneous route to cancer patients with
non-myeloid malignancies receiving chemotherapy.
Posology
Symptomatic anaemia associated
with chronic renal failure
Anaemia symptoms and sequelae may vary with age, gender, and overall burden of disease; a
physician’s evaluation of the individual patient’s clinical course and condition is necessary. Epoetin
theta should be administered either subcutaneously or intravenously in order to increase haemoglobin
level to not greater than 12 g/dl (7.45 mmol/l).
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.21 mmol/l) to 12 g/dl (7.45 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.45 mmol/l) should be avoided; guidance for appropriate dose adjustment if haemoglobin values
exceeding 12 g/dl (7.45 mmol/l) are observed are described below.
26
A rise in haemoglobin of greater than 2 g/dl (1.24 mmol/l) over a four week period should be avoided.
If the rise in haemoglobin is greater than 2 g/dl (1.24 mmol/l) in 4 weeks or the haemoglobin value
exceeds 12 g/dl (7.45 mmol/l), the dose should be reduced by 25 to 50%. It is recommended that
haemoglobin be monitored every two weeks until levels have stabilised and periodically thereafter. If
the haemoglobin level continues to increase, therapy should be interrupted until the haemoglobin level
begins to decrease, at which point therapy should be restarted at a dose approximately 25% below the
previously administered dose.
In the presence of hypertension or existing cardiovascular, cerebrovascular or peripheral vascular
diseases, the increase in haemoglobin and the target haemoglobin value should be determined
individually taking into account the clinical picture.
Treatment with epoetin theta is divided into two stages.
Correction phase
Subcutaneous administration: The initial posology is 20 IU/kg body weight 3 times per week. The
dose may be increased after 4 weeks to 40 IU/kg, 3 times per week, if the increase in haemoglobin is
not adequate (< 1 g/dl [0.62 mmol/l] within 4 weeks). Further increases of 25% of the previous dose
may be made at monthly intervals until the individual target haemoglobin level is obtained.
Intravenous administration: The initial posology is 40 IU/kg body weight 3 times per week. The dose
may be increased after 4 weeks to 80 IU/kg, 3 times per week, and by further increases of 25% of the
previous dose at monthly intervals, if needed.
For both routes of administration, the maximum dose should not exceed 700 IU/kg body weight per
week.
Maintenance phase
The dose should be adjusted as necessary to maintain the individual target haemoglobin level between
10 g/dl (6.21 mmol/l) to 12 g/dl (7.45 mmol/l), whereby a haemoglobin level of 12 g/dl (7.45 mmol/l)
should not be exceeded. If a dose adjustment is required to maintain the desired haemoglobin level, it
is recommended that the dose be adjusted by approximately 25%.
Subcutaneous administration: The weekly dose can be given as one injection per week or three times
per week.
Intravenous administration: Patients who are stable on a three times weekly dosing regimen may be
switched to twice-weekly administration.
If the frequency of administration is changed, haemoglobin level should be monitored closely and
dose adjustments may be necessary.
The maximum dose should not exceed 700 IU/kg body weight per week.
If epoetin theta is substituted for another epoetin, haemoglobin level should be monitored closely and
the same route of administration should be used.
Patients should be monitored closely to ensure that the lowest approved dose of epoetin theta is used
to provide adequate control of the symptoms of anaemia.
Symptomatic anaemia in cancer patients with non-myeloid malignancies receiving chemotherapy
Epoetin theta should be administered by the subcutaneous route to patients with anaemia (e.g.
haemoglobin concentration ≤ 10 g/dl [6.21 mmol/l]). Anaemia symptoms and sequelae may vary with
age, gender, and overall burden of disease; a physician’s evaluation of the individual patient’s clinical
course and condition is necessary.
27
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.21 mmol/l) to 12 g/dl (7.45 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.45 mmol/l) should be avoided; guidance for appropriate dose adjustment if haemoglobin values
exceeding 12 g/dl (7.45 mmol/l) are observed are described below.
The recommended initial dose is 20,000 IU, independent of bodyweight, given once-weekly. If, after
4 weeks of therapy, the haemoglobin value has increased by at least 1 g/dl (0.62 mmol/l), the current
dose should be continued. If the haemoglobin value has not increased by at least 1 g/dl (0.62 mmol/l) a
doubling of the weekly dose to 40,000 IU should be considered. If, after an additional 4 weeks of
therapy, the haemoglobin increase is still insufficient an increase of the weekly dose to 60,000 IU
should be considered.
The maximum dose should not exceed 60,000 IU per week.
If, after 12 weeks of therapy, the haemoglobin value has not increased by at least 1 g/dl (0.62 mmol/l),
response is unlikely and treatment should be discontinued.
If the rise in haemoglobin is greater than 2 g/dl (1.24 mmol/l) in 4 weeks or the haemoglobin level
exceeds 12 g/dl (7.45 mmol/l), the dose should be reduced by 25 to 50%. Treatment with epoetin theta
should be temporarily discontinued if haemoglobin levels exceed 13 g/dl (8.07 mmol/l). Therapy
should be reinitiated at approximately 25% lower than the previous dose after haemoglobin levels fall
to 12 g/dl (7.45 mmol/l) or below.
Therapy should be continued up to 4 weeks after the end of chemotherapy.
Patients should be monitored closely to ensure that the lowest approved dose of epoetin theta is used
to provide adequate control of the symptoms of anaemia.
Special populations
Paediatric patients
There is no experience in children and adolescents.
Method of administration
The solution can be administered subcutaneously or intravenously. Subcutaneous use is preferable in
patients who are not undergoing haemodialysis, in order to avoid puncturing peripheral veins. If
epoetin theta is substituted for another epoetin, the same route of administration should be used. In
cancer patients with non-myeloid malignancies receiving chemotherapy Epoetin theta should be
administered by the subcutaneous route only.
Subcutaneous injections should be given into the abdomen, arm or thigh.
Eporatio is supplied in a single use pre-filled syringe. The solution should be visually inspected prior
to use. Only clear, colourless solutions without particles should be used. The solution for injection
should not be shaken. It should be allowed to reach a comfortable temperature (15 °C - 25 °C) for
injection.
Eporatio must not be mixed with other medicinal products (see section 6.2).
The injection sites should be rotated and the injection performed slowly to avoid discomfort at the site
of injection.
-
Hypersensitivity to the active substance, other epoetins and derivatives or to any of the
excipients.
28
-
Uncontrolled hypertension.
General
Supplementary iron therapy is recommended for all patients with serum ferritin values below 100 µg/l
or with transferrin saturation below 20%. To ensure effective erythropoiesis, iron status has to be
evaluated for all patients prior to and during treatment.
Non-response to therapy with epoetin theta should prompt a search for causative factors. Deficiencies
of iron, folic acid or vitamin B
12
reduce the effectiveness of epoetins and should therefore be
corrected. Intercurrent infections, inflammatory or traumatic episodes, occult blood loss, haemolysis,
aluminium intoxication, underlying haematological diseases or bone marrow fibrosis may also
compromise the erythropoietic response. A reticulocyte count should be considered as part of the
evaluation.
Pure red cell aplasia (PRCA)
If typical causes of non-response are excluded, and the patient has a sudden drop in haemoglobin
associated with reticulocytopenia, an examination of anti-erythropoietin antibodies and the bone
marrow for diagnosis of pure red cell aplasia should be considered. Discontinuation of treatment with
epoetin theta should be taken into account.
PRCA caused by neutralising anti-erythropoietin antibodies has been reported in association with
erythropoietin therapy. These antibodies have been shown to cross-react with all epoetins, and patients
suspected or confirmed to have neutralising antibodies to erythropoietin should not be switched to
epoetin theta (see section 4.8).
A paradoxical decrease in haemoglobin and development of severe anaemia associated with low
reticulocyte counts should prompt to discontinue treatment with epoetin and perform
anti-erythropoietin antibody testing. Cases have been reported in patients with hepatitis C treated with
interferon and ribavirin, when epoetins are used concomitantly. Epoetins are not approved in the
management of anaemia associated with hepatitis C.
Hypertension
Patients on epoetin theta therapy can experience an increase in blood pressure or aggravation of
existing hypertension particularly during the initial treatment phase.
Therefore, in patients treated with epoetin theta, special care should be taken to monitor closely and
control blood pressure. Blood pressure should be controlled adequately before initiation and during
therapy to avoid acute complications, such as hypertensive crisis with encephalopathy-like symptoms
(e.g. headaches, confused state, speech disturbances, impaired gait) and related complications
(seizures, stroke), which may also occur in individual patients with otherwise normal or low blood
pressure. If these reactions occur, they require the immediate attention of a physician and intensive
medical care. Particular attention should be paid to sudden sharp migraine-like headaches as a possible
warning signal.
Increases in blood pressure may require treatment with antihypertensive medicinal products or a dose
increase of existing antihypertensive medicinal products. In addition, a reduction of the administered
dose of epoetin theta needs to be considered. If blood pressure values remain high, temporary
interruption of epoetin theta therapy may be required. Once hypertension has been controlled with
more intensified therapy, epoetin theta therapy should be re-started at a reduced dose.
Misuse
Misuse of epoetin theta by healthy persons may lead to an excessive increase in haemoglobin and
haematocrit. This may be associated with life-threatening cardiovascular complications.
29
Special populations
Due to limited experience, the efficacy and safety of epoetin theta could not be assessed in patients
with impaired liver function or homozygous sickle cell anaemia.
In clinical trials, patients over 75 years of age had a higher incidence of serious and severe adverse
events irrespective of a causal relationship to treatment with epoetin theta. Furthermore, deaths were
more frequent in this patient group compared to younger patients.
Laboratory monitoring
It is recommended that haemoglobin measurement, a complete blood count and platelet count be
performed regularly.
Symptomatic anaemia associated
with chronic renal failure
The use of epoetin theta in nephrosclerotic patients not yet undergoing dialysis should be defined
individually, as a possible accelerated progression of renal failure cannot be ruled out with certainty.
During haemodialysis, patients treated with epoetin theta may require increased anticoagulation
treatment to prevent clotting of the arterio-venous shunt.
In patients with chronic renal failure, the maintenance haemoglobin concentration should not exceed
the upper limit of the target haemoglobin concentration recommended in section 4.2. In clinical trials,
an increased risk of death and serious cardiovascular events was observed when epoetins were
administered to target a haemoglobin level in excess of 12 g/dl (7.45 mmol/l). Controlled clinical trials
have not shown significant benefits attributable to the administration of epoetins when the
haemoglobin concentration is increased beyond the level necessary to control symptoms of anaemia
and to avoid blood transfusion.
Symptomatic anaemia in cancer patients with non-myeloid malignancies receiving chemotherapy
Effect on tumour growth
Epoetins are growth factors that primarily stimulate red blood cell production. Erythropoietin
receptors may be expressed on the surface of a variety of tumour cells. As with all growth factors,
there is a concern that epoetins could stimulate the growth of any type of malignancy (see section 5.1).
In several controlled studies, epoetins have not been shown to improve overall survival or decrease the
risk of tumour progression in patients with anaemia associated with cancer. In controlled clinical
studies, use of epoetins has shown:
- shortened time to tumour progression in patients with advanced head and neck cancer receiving
radiation therapy when administered to target a haemoglobin level in excess of 14 g/dl
(8.69 mmol/l),
- shortened overall survival and increased deaths attributed to disease progression at 4 months in
patients with metastatic breast cancer receiving chemotherapy when administered to target a
haemoglobin value of 12-14 g/dl (7.45-8.69 mmol/l),
- increased risk of death when administered to target a haemoglobin value of 12 g/dl
(7.45 mmol/l) in patients with active malignant disease receiving neither chemotherapy nor
radiation therapy.
Epoetins are not indicated for use in this patient population.
In view of the above, in some clinical situations blood transfusion should be the preferred treatment
for the management of anaemia in patients with cancer. The decision to administer recombinant
erythropoietins should be based on a benefit-risk assessment with the participation of the individual
patient, which should take into account the specific clinical context. Factors that should be considered
in this assessment should include the type of tumour and its stage, the degree of anaemia,
life-expectancy, the environment in which the patient is being treated, and patient preference (see
section 5.1).
30
Excipients
This medicinal product contains less than 1 mmol sodium (23 mg) per pre-filled syringe, i.e.
essentially ‘sodium-free’.
No interaction studies have been performed.
For epoetin theta no clinical data on exposed pregnancies are available. Animal studies with other
epoetins do not indicate direct harmful effects with respect to pregnancy, embryonal/foetal
development, parturition or postnatal development (see section 5.3). Caution should be exercised
when prescribing to pregnant women.
It is unknown whether epoetin theta is excreted in human breast milk, but data in neonates show no
absorption or pharmacological activity of erythropoietin when given together with breast milk. A
decision on whether to continue/discontinue breast-feeding or to continue/discontinue therapy with
epoetin theta should be made taking into account the benefit of breast-feeding to the child and the
benefit of epoetin theta therapy for the woman.
Epoetin theta has no or negligible influence on the ability to drive and use machines.
The safety of epoetin theta has been evaluated based on results from clinical studies including
972 patients.
Approximately 9% of patients can be expected to experience an adverse reaction. The most frequent
undesirable effects are hypertension, influenza-like illness and headache.
Adverse reactions listed below are classified according to System Organ Class. Frequency groupings
are defined according to the following convention:
Very common: ≥ 1/10;
Common: ≥ 1/100 to < 1/10;
Uncommon: ≥ 1/1,000 to < 1/100;
Rare: ≥ 1/10,000 to < 1/1,000;
Very rare:
< 1/10,000;
Not known:
cannot be estimated from the available data.
31
System organ class
Adverse reaction
Frequency
Symptomatic anaemia
associated with
chronic renal failure
Symptomatic anaemia
in cancer patients with
non-myeloid
malignancies receiving
chemotherapy
Blood and lymphatic
system disorders
Thromboembolic
events
—
Not known
Shunt thrombosis
Common
—
Immune system
disorders
Hypersensitivity
reactions
Not known
Nervous system
disorders
Headache
Common
Vascular disorders
Hypertension
Common
Hypertensive crisis
Common
—
Skin and subcutaneous
tissue disorders
Skin reactions
Common
Musculoskeletal and
connective tissue
disorders
Arthralgia
—
Common
General disorders and
administration site
conditions
Influenza-like illness
Common
Shunt thrombosis may occur, especially in patients who have a tendency to hypotension or whose
arterio-venous fistulae exhibit complications (e.g. stenoses, aneurisms) (see section 4.4).
One of the most frequent adverse reactions during treatment with epoetin theta is an increase in blood
pressure or aggravation of existing hypertension particularly during the initial treatment phase.
Hypertension occurs in chronic renal failure patients more often during the correction phase than
during the maintenance phase. Hypertension can be treated with appropriate medicinal products (see
section 4.4).
Hypertensive crisis with encephalopathy-like symptoms (e.g. headaches, confused state, speech
disturbances, impaired gait) and related complications (seizures, stroke) may also occur in individual
patients with otherwise normal or low blood pressure (see section 4.4).
Skin reactions such as rash, pruritus or injection site reactions may occur.
Symptoms of influenza-like illness such as fever, chills and asthenic conditions have been reported.
Certain adverse reactions have not yet been observed with epoetin theta, but are generally accepted as
being attributable to epoetins:
In isolated cases in patients with chronic renal failure, neutralising anti-erythropoietin antibody-
mediated PRCA associated with therapy with other epoetins has been reported. If PRCA is diagnosed,
therapy with epoetin theta must be discontinued and patients should not be switched to another
recombinant epoetin (see section 4.4).
The therapeutic margin of epoetin theta is very wide. In the case of overdose, polycythaemia can
occur. In the event of polycythaemia, epoetin theta should be temporarily withheld.
If severe polycythaemia occurs, conventional methods (phlebotomy) may be indicated to reduce the
haemoglobin level.
32

















5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other anti-anaemic preparations, ATC code: B03XA01
Mechanism of action
Human erythropoietin is an endogenous glycoprotein hormone that is the primary regulator of
erythropoiesis through specific interaction with the erythropoietin receptor on the erythroid progenitor
cells in the bone marrow. It acts as a mitosis-stimulating factor and differentiation hormone. The
production of erythropoietin primarily occurs in and is regulated by the kidney in response to changes
in tissue oxygenation. Production of endogenous erythropoietin is impaired in patients with chronic
renal failure and the primary cause of their anaemia is erythropoietin deficiency. In patients with
cancer receiving chemotherapy the aetiology of anaemia is multifactorial. In these patients,
erythropoietin deficiency and a reduced response of erythroid progenitor cells to endogenous
erythropoietin both contribute significantly towards their anaemia.
Epoetin theta is identical in its amino acid sequence and similar in its carbohydrate composition
(glycosylation) to endogenous human erythropoietin.
Preclinical efficacy
The biological efficacy of epoetin theta has been demonstrated after intravenous and subcutaneous
administration in various animal models
in vivo
(mice, rats, dogs). After administration of epoetin
theta, the number of erythrocytes, the haematocrit values and reticulocyte counts increase.
Clinical efficacy and safety
Symptomatic anaemia associated chronic renal failure
Data from correction phase studies in 284 chronic renal failure patients show that the haemoglobin
response rates (defined as haemoglobin levels above 11 g/dl at two consecutive measurements) in the
epoetin theta group (88.4% and 89.4% in studies in patients on dialysis and not yet undergoing
dialysis, respectively) were comparable to epoetin beta (86.2% and 81.0%, respectively). The median
time to response was similar in the treatment groups with 56 days in haemodialysis patients and
49 days in patients not yet undergoing dialysis.
Two randomised controlled studies were conducted in 270 haemodialysis patients and 288 patients not
yet undergoing dialysis, who were on stable treatment with epoetin beta. Patients were randomised to
continue their current treatment or to be converted to epoetin theta (same dose as epoetin beta) in order
to maintain their haemoglobin levels. During the evaluation period (weeks 15 to 26), the mean and
median level of haemoglobin in patients treated with epoetin theta was virtually identical to their
baseline haemoglobin level. In these two studies, 180 haemodialysis patients and 193 patients not
undergoing dialysis were switched from maintenance phase treatment with epoetin beta to treatment
with epoetin theta for a period of six months showing stable haemoglobin values and a similar safety
profile as epoetin beta. In the clinical studies, patients not yet undergoing dialysis (subcutaneous
administration) discontinued the study more frequently than haemodialysis patients (intravenous
administration) as they had to terminate the study when starting dialysis.
In two long-term studies, the efficacy of epoetin theta was evaluated in 124 haemodialysis patients and
289 patients not yet undergoing dialysis. The haemoglobin levels remained within the desired target
range and epoetin theta was well tolerated over a period of up to 15 months.
In the clinical studies, pre-dialysis patients were treated once-weekly with epoetin theta, 174 patients
in the maintenance phase study and 111 patients in the long-term study.
Symptomatic anaemia in cancer patients with non-myeloid malignancies receiving chemotherapy
409 cancer patients receiving chemotherapy were included in two prospective, randomised
double-blind, placebo-controlled studies. The first study was conducted in 186 anaemic patients with
33
non-myeloid malignancies (55% with haematological malignancies and 45% with solid tumours)
receiving non-platinum chemotherapy. The second study was conducted in 223 patients with various
solid tumours receiving platinum-containing chemotherapy. In both studies, treatment with epoetin
theta resulted in a significant haemoglobin response (p < 0.001), defined as an increase in
haemoglobin of ≥ 2 g/dl without transfusion, and a significant reduction in transfusion requirements
(p < 0.05) in comparison to placebo.
Effect on tumour growth
Erythropoietin is a growth factor that primarily stimulates red cell production. Erythropoietin receptors
may be expressed on the surface of a variety of tumour cells.
Survival and tumour progression have been examined in five large controlled studies involving a total
of 2,833 patients, of which four were double-blind placebo-controlled studies and one was an open-
label study. Two of the studies recruited patients who were being treated with chemotherapy. The
target haemoglobin concentration in two studies was > 13 g/dl; in the remaining three studies it was
12-14 g/dl. In the open-label study there was no difference in overall survival between patients treated
with recombinant human erythropoietin and controls. In the four placebo-controlled studies the hazard
ratios for overall survival ranged between 1.25 and 2.47 in favour of controls. These studies have
shown a consistent unexplained statistically significant excess mortality in patients who have anaemia
associated with various common cancers who received recombinant human erythropoietin compared
to controls. Overall survival outcome in the trials could not be satisfactorily explained by differences
in the incidence of thrombosis and related complications between those given recombinant human
erythropoietin and those in the control group.
Data from three placebo-controlled clinical studies in 586 anaemic cancer patients conducted with
epoetin theta, showed no negative effect of epoetin theta on survival. During the studies, mortality was
lower in the epoetin theta group (6.9%) compared to placebo (10.3%).
A systematic review has also been performed involving more than 9,000 cancer patients participating
in 57 clinical trials. Meta-analysis of overall survival data produced a hazard ratio point estimate of
1.08 in favour of controls (95% CI: 0.99, 1.18; 42 trials and 8,167 patients). An increased relative risk
of thromboembolic events (RR 1.67, 95% CI: 1.35, 2.06; 35 trials and 6,769 patients) was observed in
patients treated with recombinant human erythropoietin. There is therefore consistent evidence to
suggest that there may be significant harm to patients with cancer who are treated with recombinant
human erythropoietin. The extent to which these outcomes might apply to the administration of
recombinant human erythropoietin to patients with cancer, treated with chemotherapy to achieve
haemoglobin concentrations less than 13 g/dl, is unclear because few patients with these
characteristics were included in the data reviewed.
A patient-level data analysis has also been performed on more than 13,900 cancer patients (chemo-,
radio-, chemoradio- or no therapy) participating in 53 controlled clinical trials involving several
epoetins. Meta-analysis of overall survival data produced a hazard ratio point estimate of 1.06 in
favour of controls (95% CI: 1.00, 1.12; 53 trials and 13,933 patients) and for cancer patients receiving
chemotherapy, the overall survival hazard ratio was 1.04 (95% CI: 0.97, 1.11; 38 trials and 10,441
patients). Meta-analyses also indicate consistently a significantly increased relative risk of
thromboembolic events in cancer patients receiving recombinant human erythropoietin (see section
4.4).
5.2 Pharmacokinetic properties
General
The pharmacokinetics of epoetin theta have been examined in healthy volunteers, in patients with
chronic renal failure and in cancer patients receiving chemotherapy. The pharmacokinetics of epoetin
theta are independent of age or gender.
Subcutaneous administration
34
Following subcutaneous injection of 40 IU/kg body weight epoetin theta at three different sites (upper
arm, abdomen, thigh) in healthy volunteers, similar plasma level profiles were observed. The extent of
absorption (AUC) was slightly greater after injection in the abdomen in comparison to the other sites.
The maximum concentration is reached after an average of 10 to 14 hours and the average terminal
half-life ranges from approximately 22 to 41 hours.
Average bioavailability of epoetin theta after subcutaneous administration is approximately 31%
compared with intravenous administration.
In pre-dialysis patients with chronic renal failure following subcutaneous injection of 40 IU/kg body
weight, the protracted absorption results in a concentration plateau, whereby the maximum
concentration is reached after an average of approximately 14 hours. The terminal half-life is higher
than after intravenous administration, with an average of 25 hours after single dosing and 34 hours in
steady state after repeated dosing three times weekly, without leading to an accumulation of epoetin
theta.
In cancer patients receiving chemotherapy, after repeated subcutaneous administration of 20,000 IU
epoetin theta once-weekly, the terminal half-life is 29 hours after the first dose and 28 hours in steady
state. No accumulation of epoetin theta was observed.
Intravenous administration
In patients with chronic renal failure undergoing haemodialysis, the elimination half-life of epoetin
theta is 6 hours after single dosing and 4 hours in steady state after repeated intravenous administration
of 40 IU/kg body weight epoetin theta three times weekly. No accumulation of epoetin theta was
observed. Following intravenous administration, the volume of distribution approximates to total
blood volume.
5.3 Preclinical safety data
Non-clinical data with epoetin theta reveal no special hazard for humans based on conventional studies
of safety pharmacology and repeated-dose toxicity.
Non-clinical data with other epoetins reveal no special hazard for humans based on conventional
studies of genotoxicity and toxicity to reproduction.
In reproductive toxicity studies performed with other epoetins, effects interpreted as being secondary
to decreased maternal body weight were observed at doses sufficiently in excess to the recommended
human dose.
6.
6.1 List of excipients
Sodium dihydrogen phosphate dihydrate
Sodium chloride
Polysorbate 20
Trometamol
Hydrochloric acid (6 M) (for pH adjustment)
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3 Shelf life
35
2 years.
6.4 Special precautions for storage
Store in a refrigerator (2 °C – 8 °C).
Do not freeze.
Keep the pre-filled syringe in the outer carton in order to protect from light.
For the purpose of ambulatory use, the patient may remove the product from the refrigerator and store
it at a temperature not above 25 °C for a single period of up to 7 days without exceeding the expiry
date. Once removed from the refrigerator, the medicinal product must be used within this period or
disposed of.
6.5 Nature and contents of container
0.5 ml of solution in a pre-filled syringe (type I glass) with a tip cap (bromobutyl rubber), a plunger
stopper (teflonised chlorobutyl rubber), an injection needle (stainless steel) and with or without a
pre-assembled safety device.
Pack sizes of 6 pre-filled syringes with or without safety device.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
The pre-filled syringes are for single use only.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
ratiopharm GmbH
Graf-Arco-Straße 3
89079 Ulm
Germany
info@ratiopharm.de
8.
EU/1/09/573/005
EU/1/09/573/006
9.
Date of first authorisation: 29 October 2009.
36
Detailed information on this medicinal product is available on the website of the European Medicines
Agency (EMEA)
http://www.emea.europa.eu/
.
37
1.
FURTHER INFORMATION
What Eporatio contains
-
The active substance is epoetin theta
.
Eporatio 1,000 IU/0.5 ml: One pre-filled syringe contains 1,000 international units (IU)
(8.3 micrograms) epoetin theta in 0.5 ml solution for injection corresponding to
2,000 international units (IU) (16.7 micrograms) per ml.
Eporatio 2,000 IU/0.5 ml: One pre-filled syringe contains 2,000 international units (IU)
140
-
headache (common);


-
(16.7 micrograms) epoetin theta in 0.5 ml solution for injection corresponding to
4,000 international units (IU) (33.3 micrograms) per ml.
Eporatio 3,000 IU/0.5 ml: One pre-filled syringe contains 3,000 international units (IU)
(25 micrograms) epoetin theta in 0.5 ml solution for injection corresponding to
6,000 international units (IU) (50 micrograms) per ml.
Eporatio 4,000 IU/0.5 ml: One pre-filled syringe contains 4,000 international units (IU)
(33.3 micrograms) epoetin theta in 0.5 ml solution for injection corresponding to
8,000 international units (IU) (66.7 micrograms) per ml.
Eporatio 5,000 IU/0.5 ml: One pre-filled syringe contains 5,000 international units (IU)
(41.7 micrograms) epoetin theta in 0.5 ml solution for injection corresponding to
10,000 international units (IU) (83.3 micrograms) per ml..
Eporatio 10,000 IU/1 ml: One pre-filled syringe contains 10,000 international units (IU)
(83.3 micrograms) epoetin theta in 1 ml solution for injection corresponding to
10,000 international units (IU) (83.3 micrograms) per ml.
Eporatio 20,000 IU/1 ml: One pre-filled syringe contains 20,000 international units (IU)
(166.7 micrograms) epoetin theta in 1 ml solution for injection corresponding to
20,000 international units (IU) (166.7 micrograms) per ml.
Eporatio 30,000 IU/1 ml: One pre-filled syringe contains 30,000 international units (IU)
(250 micrograms) epoetin theta in 1 ml solution for injection corresponding to
30,000 international units (IU) (250 micrograms) per ml.
What Eporatio looks like and contents of the pack
Eporatio is a solution for injection in pre-filled syringe along with an injection needle.
Eporatio 1,000 IU/0.5 ml, Eporatio 2,000 IU/0.5 ml, Eporatio 3,000 IU/0.5 ml, Eporatio
4,000 IU/0.5 ml and Eporatio 5,000 IU/0.5 ml: Each pre-filled syringe contains 0.5 ml of solution.
Packs of 6 pre-filled syringes with and without safety device.
Eporatio 10,000 IU/1 ml, Eporatio 20,000 IU/1 ml and Eporatio 30,000 IU/1 ml: Each pre-filled
syringe contains 1 ml of solution. Packs of 1, 4 or 6 pre-filled syringes with and without safety device.
Not all pack sizes may be marketed.
Eporatio is a clear and colourless solution.
Marketing Authorisation Holder
ratiopharm GmbH
Graf-Arco-Straße 3
D-89079 Ulm
Germany
info@ratiopharm.de
Manufacturer
Merckle Biotec GmbH
Dornierstraße 10
D-89079 Ulm
Germany
For any information about this medicine, please contact the local representative of the Marketing
Authorisation Holder:
België/Belgique/Belgien
ratiopharm Belgium S.A.
Tél/Tel: +32 2 761 10 11
Luxembourg/Luxemburg
ratiopharm S.A. Luxemburg
Tél/Tel: +35 2 40 37 27
141
The other ingredients are sodium dihydrogen phosphate dihydrate, sodium chloride, polysorbate
20, trometamol, hydrochloric acid (6 M) (for pH adjustment) and water for injections.
България
ratiopharm GmbH, Германия
Teл: +49 731 402 02
Magyarország
ratiopharm Hungária Kft.
Tel: +36 1 273 2730
Česká republika
ratiopharm CZ s.r.o.
Tel: +420 2 510 21 122
Malta
ratiopharm GmbH, Il Ġermanja
Tel: +49 731 402 02
Danmark
ratiopharm A/S
Tlf: +45 45 46 06 60
Nederland
ratiopharm Nederland bv
Tel: +31 23 7070 840
Deutschland
ratiopharm GmbH
Tel: +49 731 402 02
Norge
ratiopharm AS
Tlf: +47 66 77 55 90
Eesti
ratiopharm Eesti büroo
Tel: +372 6838 006
Österreich
ratiopharm Arzneimittel Vertriebs-GmbH
Tel: +43 1 97 007
Ελλάδα
ratiopharm GmbH, Γερμανία
Τηλ: +49 731 402 02
Polska
ratiopharm Polska Sp.z.o.o
Tel: +48 22 335 39 20
España
ratiopharm España, S.A.
Tel: +34 91 567 29 70
Portugal
ratiopharm, Comércio e Indústria de Produtos
farmacêuticos Lda
Tel: +351 21 424 80 00
France
Laboratoire ratiopharm S.A.
Tél: +33 1 42 07 97 04
România
ratiopharm GmbH, Germania
Tel: +49 731 402 02
Ireland
ratiopharm UK Ltd, United Kingdom
Tel: +44 239 238 6330
Slovenija
ratiopharm GmbH, Nemčija
Tel: +49 731 402 02
Ísland
ratiopharm GmbH, Þýskaland
Sími: +49 731 402 02
Slovenská republika
ratiopharm Slovensko s.r.o.
Tel: +421 2 57 200 300
Italia
ratiopharm Italia s.r.l.
Tel: +39 02 28 87 71
Suomi/Finland
ratiopharm Oy
Puh/Tel: +358 20 180 5900
Κύπρος
ratiopharm GmbH, Γερμανία
Τηλ: +49 731 402 02
Sverige
ratiopharm AB
Tel: +46 42 37 0740
Latvija
ratiopharm Latvijas pārstāvniecība
Tel: +371 67499110
United Kingdom
ratiopharm UK Ltd
Tel: +44 239 238 6330
Lietuva
ratiopharm atstovas Lietuvoje
Tel: + 370 5 212 3295
142
This leaflet was last approved in {MM/YYYY}.
INFORMATION FOR INJECTING YOURSELF
This section contains information on how to give yourself an injection of Eporatio under the skin. It is
important that you do not try to give yourself the injection unless you have received special training
from your doctor or nurse. If you are not sure about giving yourself the injection or you have any
questions, please ask your doctor or nurse for help.
How do I use Eporatio?
You will need to give yourself the injection into the tissue just under the skin. This is known as a
subcutaneous injection.
Equipment that I need
To give yourself an injection into the tissue under the skin you will need:
-
a pre-filled syringe of Eporatio,
-
an alcohol wipe,
-
a piece of gauze bandage or a sterile gauze swab,
-
a puncture-proof container (plastic container provided by the hospital or pharmacy) so you can
dispose of used syringes safely.
What should I do before my injection?
Eporatio is supplied in pre-filled syringes either with or without safety device. The following figures
refer either to syringes without (Figures 2a, 3a, 4a) or with (Figures 2b, 3b, 4b, 9) safety device. The
other figures (1, 5, 6, 7, 8) apply to both syringes. Your doctor or pharmacist can tell you which
pre-filled syringe you have received.
1. Take one blister with an Eporatio pre-filled syringe out of the refrigerator.
2. Take the pre-filled syringe and the needle container out of the blister.
3. Check the expiry date on the pre-filled syringe label (EXP). Do not use it if the date has passed
the last day of the month shown.
4. Check the appearance of Eporatio. It must be a clear and colourless liquid. If there are particles
in it or if it is cloudy, you must not use it.
5. There is a cap at the end of the needle container. Break the labelled seal and remove the cap (see
picture 1).
6. Remove the tip cap from the pre-filled syringe (see picture 2a for syringes without safety device
and picture 2b for syringes with safety device).
7. Attach the needle to the syringe (see picture 3a for syringes without safety device and picture 3b
for syringes with safety device). Do not remove the needle cover at this time.
8. For a more comfortable injection, let the pre-filled syringe stand for 30 minutes to reach room
temperature or hold the pre-filled syringe gently in your hand for a few minutes. Do not warm
Eporatio in any other way (for example, do
not
warm it in a microwave or in hot water).
9. Do
not
remove the needle cover from the syringe until you are ready to inject.
10.
Wash your hands thoroughly.
11. Find a comfortable, well-lit place. Put everything you need where you can reach them (the
Eporatio pre-filled syringe, an alcohol wipe, a piece of gauze bandage or a sterile gauze swab
and the puncture-proof container).
143

1
Without safety device
2a
2b
With safety device
3a
Without safety device
144



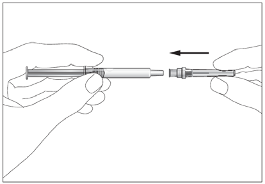
3b
With safety device
How do I prepare my injection?
Before you give yourself an Eporatio injection, you must do the following:
1.
Hold the syringe and gently remove the cover from the needle without twisting. Pull straight as
shown in picture 4a for syringes without safety device and picture 4b for syringes with safety
device. Do not touch the needle or push the plunger.
2.
You may notice a small air bubble in the pre-filled syringe. If there are air bubbles present,
gently tap the syringe with your fingers until the air bubbles rise to the top of the syringe. With
the syringe pointing upwards, expel all air from the syringe by pushing the plunger slowly
upwards.
3.
The syringe has a scale on the syringe barrel. Push the plunger up to the number (IU) on the
syringe that matches the dose of Eporatio that your doctor prescribed.
4.
Check again to make sure the correct dose of Eporatio is in the syringe.
4a
Without safety device
With safety device
4b
Where should I give my injection?
The most suitable places to inject yourself are:
-
the top of your thighs; and
145
5.
You can now use the pre-filled syringe.
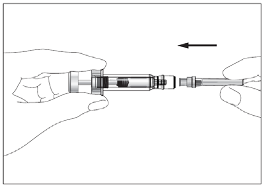

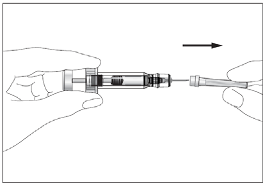
-
the abdomen, except for the area around the navel (see grey areas in picture 5).
If someone else is injecting you, they can also use the back of your arms (see grey areas in picture 6).
It is better to change the injection site every day to avoid the risk of soreness at any one site.
5
6
How do I give my injection?
1.
Disinfect the injection site on the skin by using an alcohol wipe and pinch the skin between
your thumb and forefinger, without squeezing it (see picture 7).
2.
Put the needle fully into the skin as shown by your nurse or doctor. The angle between the
syringe and skin should not be too narrow (≥ 45°, see picture 8).
3.
Pull slightly on the plunger to check that a blood vessel has not been punctured. If you see
blood in the syringe, remove the needle and re-insert it in another place.
4.
Inject the liquid into the tissue slowly and evenly, always keeping your skin pinched.
5.
Syringe without safety device: After injecting the liquid, remove the needle and let go of your
skin.
Syringe with safety device: Do not release the plunger fully until after the full dose has been
given. The entire needle will be removed automatically from the skin and safely guarded so that
you can not prick yourself (see picture 9).
6.
Press the injection site with a piece of gauze bandage or a sterile gauze swab for several
seconds.
7.
Only use each syringe for one injection. Do not use any Eporatio that is left in the syringe.
146
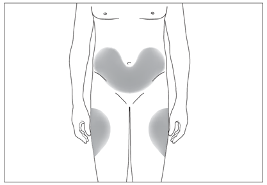
7
8
With safety device
9
Remember
If you have any problems, please do not be afraid to ask your doctor or nurse for help and advice.
Disposing of used syringes
-
Do not put the cover back on used needles.
-
Put used syringes into the puncture-proof container and keep this container out of the reach and
sight of children.
-
Dispose of the full puncture-proof container as instructed by your doctor, nurse or pharmacist.
-
Never put the syringes that you have used into your normal household rubbish bin.
147
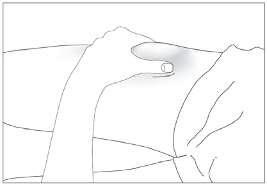


Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/eporatio.html
Copyright © 1995-2021 ITA all rights reserved.