










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
IRESSA 250 mg film-coated tablets
2.
Each tablet contains 250 mg of gefitinib.
Excipient: Each tablet contains 163.5 mg of lactose (as monohydrate)
For a full list of excipients, see section 6.1.
3.
Film-coated tablets (tablet).
Tablets are brown, round, biconvex, impressed with “IRESSA 250” on one side and plain on the other.
4.
IRESSA is indicated for the treatment of adult patients with locally advanced or metastatic non-small
cell lung cancer (NSCLC) with activating mutations of EGFR-TK (see section 5.1).
Treatment with IRESSA should be initiated and supervised by a physician experienced in the use of
anticancer therapies.
Posology
The recommended posology of IRESSA is one 250 mg tablet once a day. If a dose of IRESSA is
missed, it should be taken as soon as the patient remembers. If it is less than 12 hours to the next dose,
the patient should not take the missed dose. Patients should not take a double dose (two doses at the
same time) to make up for a forgotten dose.
Paediatric population
The safety and efficacy of IRESSA in children and adolescents aged less than 18 years have not been
established. There is no relevant use of IRESSA in the paediatric population in the indication of
NSCLC.
Hepatic impairment
Patients with moderate to severe hepatic impairment (Child Pugh B or C) due to cirrhosis have
increased plasma concentrations of gefitinib. These patients should be closely monitored for adverse
events. Plasma concentrations were not increased in patients with elevated aspartate transaminase
(AST), alkaline phosphatase or bilirubin due to liver metastases (see section 5.2).
Renal impairment
No dose adjustment is required in patients with impaired renal function at creatinine clearance
>20 ml/min. Only limited data are available in patients with creatinine clearance ≤ 20 ml/min and
caution is advised in these patients (see section 5.2 ).
Elderly
No dose adjustment is required on the basis of patient age (see section 5.2)
2
CYP2D6 poor metabolisers
No specific dose adjustment is recommended in patients with known CYP2D6 poor metaboliser
genotype, but these patients should be closely monitored for adverse events (see section 5.2).
Dose adjustment due to toxicity
Patients with poorly tolerated diarrhoea or skin adverse reactions may be successfully managed by
providing a brief (up to 14 days) therapy interruption followed by reinstatement of the 250 mg dose
(see section 4.8). For patients unable to tolerate treatment after a therapy interruption, IRESSA should
be discontinued and an alternative treatment should be considered.
Method of administration
The tablet may be taken with or without food, at about the same time each day. The tablet can be
swallowed whole with some water or if dosing of whole tablets is not possible, tablets may be
administered as a dispersion in water (non-carbonated). No other liquids should be used. Without
crushing it, the tablet should be dropped in half a glass of drinking water. The glass should be swirled
occasionally, until the tablet is dispersed (this may take up to 20 minutes). The dispersion should be
drunk immediately after dispersion is complete (i.e. within 60 minutes). The glass should be rinsed
with half a glass of water, which should also be drunk. The dispersion can also be administered
through a naso-gastric or gastrostomy tube.
Hypersensitivity to the active substance or to any of the excipients.
Breast-feeding (see section 4.6)
Assessment of EGFR mutation status
When assessing the EGFR mutation status of a patient, it is important that a well-validated and robust
methodology is chosen to avoid false negative or false positive determinations.
Interstitial lung disease (ILD)
ILD, which may be acute in onset, has been observed in 1.3 % of patients receiving IRESSA, and
some cases have been fatal (see section 4.8). If patients experience worsening of respiratory symptoms
such as dyspnoea, cough and fever, IRESSA should be interrupted and the patient should be promptly
investigated. If ILD is confirmed, IRESSA should be discontinued and the patient treated
appropriately.
In a Japanese pharmacoepidemiological case control study in 3159 patients with NSCLC receiving
IRESSA or chemotherapy who were followed up for 12 weeks, the following risk factors for
developing ILD (irrespective of whether the patient received IRESSA or chemotherapy) were
identified: smoking, poor performance status (PS≥ 2), CT scan evidence of reduced normal lung (≤
50 %), recent diagnosis of NSCLC (< 6 months), pre-existing ILD, older age (≥ 55 years old) and
concurrent cardiac disease. An increased risk of ILD on gefitinib relative to chemotherapy was seen
predominantly during the first 4 weeks of treatment (adjusted OR 3.8; 95 % CI 1.9 to 7.7); thereafter
the relative risk was lower (adjusted OR 2.5; 95 % CI 1.1 to 5.8). Risk of mortality among patients
who developed ILD on IRESSA or chemotherapy was higher in patients with the following risk
factors: smoking, CT scan evidence of reduced normal lung (≤ 50 %), pre-existing ILD, older age (≥
65 years old), and extensive areas adherent to pleura (≥ 50 %).
Hepatotoxicity and liver impairment
Liver function test abnormalities (including increases in
alanine aminotransferase, aspartate
aminotransferase, bilirubin) have been observed, uncommonly presenting as hepatitis (see section 4.8).
There have been isolated reports of hepatic failure which in some cases led to fatal outcomes.
Therefore, periodic liver function testing is recommended. IRESSA should be used cautiously in the
3
presence of mild to moderate changes in liver function. Discontinuation should be considered if
changes are severe.
Impaired liver function due to cirrhosis has been shown to lead to increased plasma concentrations of
gefitinib (see section 5.2).
Interactions with other medicinal products
CYP3A4 inducers may increase metabolism of gefitinib and decrease gefitinib plasma concentrations.
Therefore, concomitant administration of CYP3A4 inducers (e.g. phenytoin, carbamazepine,
rifampicin, barbiturates or herbal preparations containing St John’s wort/
Hypericum perforatum
) may
reduce efficacy of the treatment and should be avoided (see section 4.5).
In individual patients with CYP2D6 poor metaboliser genotype, treatment with a potent CYP3A4
inhibitor might lead to increased plasma levels of gefitinib. At initiation of treatment with a CYP3A4
inhibitor, patients should be closely monitored for gefitinib adverse reactions (see section 4.5).
International normalised ratio (INR) elevations and/or bleeding events have been reported in some
patients taking warfarin together with gefitinib (see section 4.5). Patients taking warfarin and gefitinib
concomitantly should be monitored regularly for changes in prothrombin time (PT) or INR.
Medicinal products that cause significant sustained elevation in gastric pH, such as proton-pump
inhibitors and h
2
-antagonists may reduce bioavailability and plasma concentrations of gefitinib and,
therefore, may reduce efficacy. Antacids if taken regularly close in time to administration of IRESSA
may have a similar effect (see sections 4.5 and 5.2).
Data from phase II clinical trials, where gefitinib and vinorelbine have been used concomitantly,
indicate that gefitinib may exacerbate the neutropenic effect of vinorelbine.
Lactose
IRESSA contains lactose. Patients with rare hereditary problems of galactose intolerance, the Lapp
lactose deficiency or glucose-galactose malabsorption should not take this medicinal product.
Further precautions for use
Patients should be advised to seek medical advice immediately if they experience:
• any eye symptoms.
• severe or persistent diarrhoea, nausea, vomiting or anorexia as these may indirectly lead to
dehydration.
These symptoms should be managed as clinically indicated (see section 4.8).
In a phase I/II trial studying the use of gefitinib and radiation in paediatric patients, with newly
diagnosed brain stem glioma or incompletely resected supratentorial malignant glioma, 4 cases (1
fatal) of Central Nervous System (CNS) haemorrhages were reported from 45 patients enrolled. A
further case of CNS haemorrhage has been reported in a child with an ependymoma from a trial with
gefitinib alone. An increased risk of cerebral haemorrhage in adult patients with NSCLC receiving
gefitinib has not been established.
Gastrointestinal perforation has been reported in patients taking IRESSA. In most cases this is
associated with other known risk factors, including concomitant medications such as steroids or
NSAIDS, underlying history of GI ulceration, age, smoking or bowel metastases at sites of
perforation.
The metabolism of gefitinib is via the cytochrome P450 isoenzyme CYP3A4 (predominantly) and via
CYP2D6.
Active substances that may increase gefitinib plasma concentrations
4
In vitro
studies have shown that gefitinib is a substrate of p-glycoprotein (Pgp). Available data do not
suggest any clinical consequences to this
in vitro
finding.
Substances that inhibit CYP3A4 may decrease the clearance of gefitinib. Concomitant administration
with potent inhibitors of CYP3A4 activity (e.g. ketoconazole, posaconazole, voriconazole, protease
inhibitors, clarithromycin, telithromycin) may increase gefitinib plasma concentrations. The increase
may be clinically relevant since adverse reactions are related to dose and exposure. The increase
might be higher in individual patients with CYP2D6 poor metaboliser genotype. Pre-treatment with
itraconazole (a potent CYP3A4 inhibitor) resulted in an 80 % increase in the mean AUC of gefitinib in
healthy volunteers. In situations of concomitant treatment with potent inhibitors of CYP3A4 the
patient should be closely monitored for gefitinib adverse reactions.
There are no data on concomitant treatment with an inhibitor of CYP2D6 but potent inhibitors of this
enzyme might cause increased plasma concentrations of gefitinib in CYP2D6 extensive metabolisers
by about 2-fold (see section 5.2). If concomitant treatment with a potent CYP2D6 inhibitor is initiated,
the patient should be closely monitored for adverse reactions.
Active substances that may reduce gefitinib plasma concentrations
Substances that are inducers of CYP3A4 activity may increase metabolism and decrease gefitinib
plasma concentrations and thereby reduce the efficacy of IRESSA. Concomitant medicinal products
that induce CYP3A4 (e.g. phenytoin, carbamazepine, rifampicin, barbiturates or St John’s wort
(
Hypericum perforatum
)), should be avoided. Pre-treatment with rifampicin (a potent CYP3A4
inducer) in healthy volunteers reduced mean gefitinib AUC by 83 % (see section 4.4).
Substances that cause significant sustained elevation in gastric pH may reduce gefitinib plasma
concentrations and thereby reduce the efficacy of IRESSA. High doses of short-acting antacids may
have a similar effect if taken regularly close in time to administration of gefitinib. Concomitant
administration of gefitinib with ranitidine at a dose that caused sustained elevations in gastric pH ≥5,
resulted in a reduced mean gefitinib AUC by 47 % in healthy volunteers (see section 4.4 and 5.2).
Active substances that may have their plasma concentrations altered by gefitinib
In vitro
studies have shown that gefitinib has limited potential to inhibit CYP2D6. In a clinical trial in
patients, gefitinib was co-administered with metoprolol (a CYP2D6 substrate). This resulted in a 35 %
increase in exposure to metoprolol. Such an increase might potentially be relevant for CYP2D6
substrates with narrow therapeutic index. When the use of CYP2D6 substrates are considered in
combination with gefitinib, a dose modification of the CYP2D6 substrate should be considered
especially for products with a narrow therapeutic window.
Gefitinib inhibits the transporter protein BCRP
in vitro
, but the clinical relevance of this finding is
unknown.
Other potential interactions
INR elevations and/or bleeding events have been reported in some patients concomitantly taking
warfarin (see section 4.4).
Women of childbearing potential
Women of childbearing potential must be advised not to get pregnant during therapy.
Pregnancy
There are no data from the use of gefitinib in pregnant women. Studies in animals have shown
reproductive toxicity (see section 5.3). The potential risk for humans is unknown. IRESSA should not
be used during pregnancy unless clearly necessary.
5
Breastfeeding
It is not known whether gefitinib is secreted in human milk. Gefitinib and metabolites of gefitinib
accumulated in milk of lactating rats (see section 5.3). IRESSA is contraindicated during
breast-feeding and therefore breast-feeding must be discontinued while receiving IRESSA therapy
(see section 4.3).
IRESSA has no or negligible influence on the ability to drive and use machines.
However, during treatment with gefitinib, asthenia has been reported. Therefore, patients who
experience this symptom should be cautious when driving or using machines.
In the pooled dataset from the ISEL, INTEREST and IPASS phase III clinical trials (2462
IRESSA-treated patients), the most frequently reported adverse drug reactions (ADRs), occurring in
more than 20 % of the patients, are diarrhoea and skin reactions (including rash, acne, dry skin and
pruritus). ADRs usually occur within the first month of therapy and are generally reversible.
Approximately 8 % of patients had a severe ADR (common toxicity criteria, (CTC) grade 3 or 4).
Approximately 3 % of patients stopped therapy due to an ADR.
Interstitial lung disease (ILD) has occurred in 1.3 % of patients, often severe (CTC grade 3-4). Cases
with fatal outcomes have been reported.
The safety profile presented in Table 1 is based on the gefitinib clinical development programme and
postmarketed experience. Adverse reactions have been assigned to the frequency categories in Table
1 where possible based on the incidence of comparable adverse event reports in a pooled dataset from
the ISEL, INTEREST and IPASS phase III clinical trials (2462 IRESSA-treated patients).
Frequencies of occurrence of undesirable effects are defined as: very common (≥ 1/10); common
(> 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare
(< 1/10,000), not known (cannot be estimated from the available data).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Table 1 Adverse reactions
Adverse reactions by system organ class and frequency
Metabolism and nutrition
disorders
Very Common
Anorexia mild or moderate
(CTC grade 1 or 2).
Eye disorders
Common
Conjunctivitis, blepharitis, and
dry eye*, mainly mild (CTC
grade 1).
Uncommon
Corneal erosion, reversible and
sometimes in association with
aberrant eyelash growth.
Vascular disorders
Common
Haemorrhage, such as epistaxis
and haematuria.
Respiratory, thoracic and
mediastinal disorders
Common
Interstitial lung disease (1.3 %),
often severe (CTC grade 3-4).
Cases with fatal outcomes have
been reported.
Gastrointestinal disorders
Very Common
Diarrhoea, mainly mild or
moderate (CTC grade 1 or 2).
6











Vomiting, mainly mild or
moderate (CTC grade 1 or 2).
Nausea, mainly mild (CTC
grade 1).
Common
Stomatitis, predominantly mild
in nature (CTC grade 1).
Dehydration, secondary to
diarrhoea, nausea, vomiting or
anorexia.
Dry mouth*, predominantly
mild (CTC grade 1).
Uncommon
Pancreatitis; gastrointestinal
perforation
Hepatobiliary disorders
Very Common
Elevations in alanine
aminotransferase, mainly mild
to moderate.
Elevations in aspartate
aminotransferase, mainly mild
to moderate.
Common
Elevations in total bilirubin,
mainly mild to moderate.
Uncommon
Hepatitis***
Skin and subcutaneous tissue
disorders
Very Common
Skin reactions, mainly a mild or
moderate (CTC grade 1 or 2)
pustular rash, sometimes itchy
with dry skin, including skin
fissures, on an erythematous
base.
Nail disorder
Common
Alopecia
Uncommon
Allergic reactions**, including
angioedema and urticaria
Rare
Bullous conditions including
Toxic epidermal necrolysis,
Stevens Johnson syndrome and
erythema multiforme
Renal and urinary disorders
Common
Cutaneous vasculitis
Asymptomatic laboratory
elevations in blood creatinine
Proteinuria
Cystitis
Rare
Haemorrhagic cystitis
General disorders
Very Common
Asthenia, predominantly mild
(CTC grade 1).
Common
Pyrexia
Frequency of ADRs relating to abnormal laboratory values is based on patients with a change in baseline
of 2 or more CTC grades in the relevant laboratory parameters.
7



































*This event can occur in association with other dry conditions (mainly skin reactions) seen with IRESSA.
**The overall incidence of adverse events of allergic reaction reported in the pooled analysis of the ISEL,
INTEREST and IPASS trials was 1.5 % (36 patients). Fourteen of the 36 patients were excluded from the
reported frequency as their reports contained evidence of either a non allergic aetiology or that the allergic
reaction was the result of treatment with another medicinal product.
***This includes isolated reports of hepatic failure which in some cases led to fatal outcomes.
Interstitial lung disease (ILD)
In the INTEREST trial, the incidence of ILD type events was 1.4 % (10) patients in the gefitinib group
vs.
1.1 % (8) patients in the docetaxel group. One ILD-type event was fatal, and this occurred in a
patient receiving gefitinib.
In the ISEL trial, the incidence of ILD-type events in the overall population was approximately 1 % in
both treatment arms. The majority of ILD-type events reported was from patients of Asian ethnicity
and the ILD incidence among patients of Asian ethnicity receiving gefitinib therapy and placebo was
approximately 3 % and 4 % respectively. One ILD-type event was fatal, and this occurred in a patient
receiving placebo.
In a post-marketing surveillance study in Japan (3350 patients) the reported rate of ILD-type events in
patients receiving gefitinib was 5.8 %. The proportion of ILD-type events with a fatal outcome was
38.6 %.
In a phase III open-label clinical trial (IPASS) in 1217 patients comparing IRESSA to
carboplatin/paclitaxel doublet chemotherapy as first-line treatment in selected patients with advanced
NSCLC in Asia, the incidence of ILD-type events was 2.6 % on the IRESSA treatment arm versus
1.4 % on the carboplatin/paclitaxel treatment arm.
There is no specific treatment in the event of overdose of gefitinib. However, in phase I clinical trials,
a limited number of patients were treated with daily doses of up to 1000 mg. An increase of frequency
and severity of some adverse reactions was observed, mainly diarrhoea and skin rash. Adverse
reactions associated with overdose should be treated symptomatically; in particular severe diarrhoea
should be managed as clinically indicated. In one study a limited number of
patients were treated
weekly
with doses from 1500 mg
to
3500 mg
.
In this study IRESSA exposure did not increase with
increasing dose, adverse events were mostly mild to moderate in severity, and were consistent with the
known safety profile of IRESSA.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Protein kinase inhibitors; ATC code: L01XE02
Mechanism of action and pharmacodynamic effects
The epidermal growth factor (EGF) and its receptor (EGFR [HER1; ErbB1]) have been identified as
key drivers in the process of cell growth and proliferation for normal and cancer cells. EGFR
activating mutation within a cancer cell is an important factor in promotion of tumour cell growth,
blocking of apoptosis, increasing the production of angiogenic factors and facilitating the processes of
metastasis.
Gefitinib is a selective small molecule inhibitor of the epidermal growth factor receptor tyrosine
kinase and is an effective treatment for patients with tumours with activating mutations of the EGFR
tyrosine kinase domain regardless of line of therapy. No clinically relevant activity has been shown in
patients with known EGFR mutation-negative tumours.
8
Clinical efficacy and safety
First line treatment
The randomised phase III first line IPASS study
was conducted in patients in Asia
1
with advanced
(stage IIIB or IV) NSCLC of adenocarcinoma histology who were ex-light smokers (ceased smoking
>
15 years ago and smoked
<
10 pack years) or never smokers (see Table 2).
1
China, Hong Kong, Indonesia, Japan, Malaysia, Philippines, Singapore, Taiwan and Thailand.
Table 2 Efficacy outcomes for gefitinib versus carboplatin/paclitaxel from the IPASS study
Population
N
Objective response
rates and 95
% CI
for difference
between
treatments
a
Primary endpoint
Progression free survival
ab
Overall survival
abc
Overall
1217
43.0
% vs 32.2
%
[5.3
%, 16.1
%]
HR 0.74
[0.65, 0.85]
5.7
m vs 5.8
m
p<0.0001
HR 0.91
[0.76, 1.10]
18.6
m vs 17.
3m
EGFR
mutation-positive
261
71.2
% vs 47.3
%
[12.0
%, 34.9
%]
HR 0.48
[0.36, 0.64]
9.5
m vs 6.3
m
p<0.0001
HR 0.78
[0.50, 1.20]
NR vs 19.5
m
EGFR
mutation-negative
176
1.1
% vs 23.5
%
[-32.5
%, -13.3
%]
HR 2.85
[2.05, 3.98]
1.5
m vs 5.5
m
p<0.0001
HR 1.38
[0.92, 2.09]
12.1
m vs 12.6
m
a Values presented are for IRESSA versus carboplatin/paclitaxel.
b “m” is medians in months. Numbers in square brackets are 95 % confidence intervals for HR
c From early analysis, overall survival follow up is ongoing
NR Not reached
N Number of patients randomised.
HR Hazard ratio (hazard ratios <1 favour IRESSA)
Quality of life outcomes differed according to EGFR mutation status. In EGFR mutation-positive
patients, significantly more IRESSA-treated patients experienced an improvement in quality of life
and lung cancer symptoms vs carboplatin/paclitaxel (see Table 3).
Table 3 Quality of life outcomes for gefitinib versus carboplatin/paclitaxel from the IPASS study
Population
N
FACT-L QoL improvement rate
a
%
LCS symptom improvement rate
a
%
Overall
1151
(48.0
% vs 40.8
%)
p=0.0148
(51.5
% vs 48.5
%)
p=0.3037
EGFR mutation-positive 259
(70.2
% vs 44.5
%)
p<0.0001
(75.6
% vs 53.9
%)
p=0.0003
9








Population
N
FACT-L QoL improvement rate
a
%
LCS symptom improvement rate
a
%
EGFR
mutation-negative
169
(14.6
% vs 36.3
%)
p=0.0021
(20.2
% vs 47.5
%)
p=0.0002
Trial outcome index results were supportive of FACT-L and LCS results
a
Values presented are for IRESSA versus carboplatin/paclitaxel.
N Number of patients evaluable for quality of life analyses
QoL Quality of life
FACT-L Functional assessment of cancer therapy-lung
LCS Lung cancer subscale
Pretreated Patients
The randomised phase III INTEREST study was conducted in patients with locally advanced or
metastatic NSCLC who had previously received platinum-based chemotherapy. In the overall
population, no statistically significant difference between gefitinib and docetaxel (75 mg/m2) was
observed for overall survival, progression free survival and objective response rates (see table 4).
Table 4 Efficacy outcomes for gefitinib versus docetaxel from the INTEREST study
Population
N
Objective response
rates and 95
% CI
for difference
between
treatments
a
Progression free survival
ab
Primary endpoint
overall survival
ab
Overall
1466
9.1
% vs 7.6
%
[-1.5
%, 4.5
%]
HR 1.04
[0.93,1.18]
2.2
m vs 2.7
m
p=0.4658
HR 1.020
[0.905, 1.150]
c
7.6
m vs 8.0
m
p=0.7332
EGFR
mutation-positive
44
42.1
% vs 21.1
%
[-8.2
%, 46.0
%]
HR 0.16
[0.05, 0.49]
7.0
m vs 4.1
m
p=0.0012
HR 0.83
[0.41, 1.67]
14.2
m vs 16.6
m
p=0.6043
EGFR mutation-
negative
253
6.6
% vs 9.8
%
[-10.5
%, 4.4
%]
HR 1.24
[0.94,1.64]
1.7
m vs 2.6
m
p=0.1353
HR 1.02
[0.78, 1.33]
6.4
m vs 6.0
m
p=0.9131
Asians
c
323
19.7
% vs 8.7
%
[3.1
%, 19.2
%]
HR 0.83
[0.64,1.08]
2.9
m vs 2.8
m
p=0.1746
HR 1.04
[0.80, 1.35]
10.4
m vs 12.2
m
p=0.7711
Non-Asians
1143
6.2
% vs 7.3
%
[-4.3
%, 2.0
%]
HR 1.12
[0.98, 1.28]
2.0
m vs 2.7
m
p=0.1041
HR 1.01
[0.89, 1.14]
6.9
m vs 6.9
m
p=0.9259
a Values presented are for IRESSA versus docetaxel.
b “m” is medians in months. Numbers in square brackets are 96 % confidence interval for overall survival
HR in the overall population, or otherwise 95 % confidence intervals for HR
c Confidence interval entirely below non-inferiority margin of 1.154
N Number of patients randomised.
10









HR Hazard ratio (hazard ratios <1 favour IRESSA)
Figures 1 and 2 Efficacy outcomes in subgroups of non-Asian patients in the INTEREST study
(N patients = Number of patients randomised)
Overall Survival
N patients
1143
Overall
27
EGFR Mutation+
222
EGFR Mutation-
133
Never-smoker
1010
Ever-smoker
600
Adenocarcinoma
543
Non-adenocarcinoma
369
Female
774
Male
0.5
1.0
1.5
2.0
Hazard Ratio (Gefitinib versus Docetaxel) and 95% CI
Unadjusted analysis PP population for clinical factors ITT population for biomarker factors
Progression-free Survival
N patients
ORR (%)
Gefitinib v. Docetaxel
1143
6.2 v. 7.3
Overall
27
42.9 v. 20.0
EGFR Mutation+
222
5.5 v. 9.1
EGFR Mutation
133
1010
23.7 v. 13.3
Never-smoker
3.9 v. 6.5
Ever-smoker
600
543
9.4 v. 9.4
Adenocarcinoma
2.8 v. 5.0
Non-adenocarcinoma
369
9.8 v. 13.1
Female
774
4.4 v. 4.6
Male
0
0.5
1.0
1.5
2.0
Hazard Ratio (Gefitinib versus Docetaxel) and 95% CI
Unadjusted analysis EFR population
The randomised phase III ISEL study, was conducted in patients with advanced NSCLC who had
received 1 or 2 prior chemotherapy regimens and were refractory or intolerant to their most recent
regimen. Gefitinib plus best supportive care was compared to placebo plus best supportive care.
IRESSA did not prolong survival in the overall population. Survival outcomes differed by smoking
status and ethnicity (see Table 5).
11

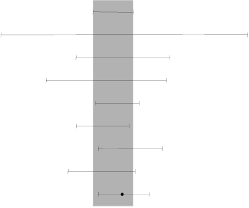









































Table 5 Efficacy outcomes for gefitinib versus placebo from the ISEL study
Population
N
Objective response
rates and 95
% CI
for difference
between
treatments
a
Time to treatment failure
ab
Primary endpoint
overall survival
abc
Overall
1692
8.0
% vs 1.3
%
[4.7
%, 8.8
%]
HR 0.82
[0.73, 0.92]
3.0
m vs 2.6
m
p=0.0006
HR 0.89
[0.77,1.02]
5.6
m vs 5.1
m
p=0.0871
EGFR mutation-
positive
26
37.5
% vs 0
%
[-15.1
%, 61.4
%]
HR 0.79
[0.20, 3.12]
10.8
m vs 3.8m
p=0.7382
HR NC
NR vs 4.3
m
EGFR mutation-
negative
189
2.6
% vs 0
%
[-5.6
%, 7.3
%]
HR 1.10
[0.78, 1.56]
2.0
m vs 2.6
m
p=0.5771
HR 1.16
[0.79, 1.72]
3.7
m vs 5.9
m
p=0.4449
Never smoker
375
18.1
% vs 0
%
[12.3
%, 24.0
%]
HR 0.55
[0.42, 0.72]
5.6
m vs 2.8
m
p<0.0001
HR 0.67
[0.49, 0.92]
8.9
m vs 6.1
m
p=0.0124
Ever smoker
1317
5.3
% vs 1.6
%
[1.4
%, 5.7
%]
HR 0.89
[0.78, 1.01]
2.7
m vs 2.6
m
p=0.0707
HR 0.92
[0.79, 1.06]
5.0
m vs 4.9
m
p=0.2420
Asians
d
342
12.4
% vs 2.1
%
[4.0
%, 15.8
%]
HR 0.69
[0.52, 0.91]
4.4
m vs 2.2
m
p=0.0084
HR 0.66
[0.48, 0.91]
9.5
m vs 5.5
m
p=0.0100
Non-Asians
1350
6.8
% vs 1.0
%
[3.5
%, 7.9
%]
HR 0.86
[0.76, 0.98]
2.9
m vs 2.7
m
p=0.0197
HR 0.92
[0.80, 1.07]
5.2
m vs 5.1
m
p=0.2942
a Values presented are for IRESSA versus placebo.
b “m” is medians in months. Numbers in square brackets are 95 % confidence intervals for HR
c Stratified log-rank test for overall; otherwise cox proportional hazards model
d Asian ethnicity excludes patients of Indian origin and refers to the racial origin of a patient group and not
necessarily their place of birth
N Number of patients randomised
NC Not calculated for overall survival HR as the number of events is too few
NR Not reached
HR Hazard ratio (hazard ratios <1 favour IRESSA)
EGFR mutation status and clinical characteristics
Clinical characteristics of never smoker, adenocarcinoma histology, and female gender have been
shown to be independent predictors of positive EGFR mutation status in a multivariate analysis of 786
Caucasian patients from gefitinib studies* (see Table 6). Asian patients also have a higher incidence
of EGFR mutation-positive tumours.
12








Table 6 Summary of multivariate logistic regression analysis to identify factors that independently
predicted for the presence of EGFR mutations in 786 Caucasian patients*
Factors that
predicted for
presence of
EGFR mutation
p-value
Odds of EGFR
mutation
Positive predictive value (9.5 % of the
overall population are EGFR
mutation-positive (M+))
Smoking status
<0.0001 6.5 times higher in never
smokers than
ever-smokers
28/70 (40 %) of never smokers are M+
47/716 (7 %) of ever smokers are M+
Histology
<0.0001 4.4 times higher in
adenocarcinoma than in
non-adenocarcinoma
63/396 (16 %) of patients with
adenocarcinoma histology are M+
12/390 (3 %) of patients with
non-adenocarcinoma histology are M+
40/235 (17 %) of females are M+
35/551 (6 %) of males are M+
*from the following studies: INTEREST, ISEL, INTACT 1&2, IDEAL 1&2, INVITE
0.0397
1.7 times higher in
females than males
5.2 Pharmacokinetic properties
Absorption
Following oral administration of gefitinib, absorption is moderately slow and peak plasma
concentrations of gefitinib typically occur at 3 to 7 hours after administration. Mean absolute
bioavailability is 59 % in cancer patients. Exposure to gefitinib is not significantly altered by food. In a
trial in healthy volunteers where gastric pH was maintained above pH 5, gefitinib exposure was
reduced by 47 %, likely due to impaired solubility of gefitinib in the stomach (see sections 4.4 and
4.5).
Distribution
Gefitinib has a mean steady state volume of distribution of 1400 l indicating extensive distribution into
tissue. Plasma protein binding is approximately 90 %. Gefitinib binds to serum albumin and alpha
1-acid glycoprotein.
In vitro
data indicate that gefitinib is a substrate for the membrane transport protein Pgp.
Metabolism
In vitro
data indicate that CYP3A4 and CYP2D6 are the major P450 isozyme involved in the
oxidative metabolism of gefitinib.
In vitro
studies have shown that gefitinib has limited potential to inhibit CYP2D6. Gefitinib shows no
enzyme induction effects in animal studies and no significant inhibition (
in vitro
) of any other
cytochrome P450 enzyme.
Gefitinib is extensively metabolised in humans. Five metabolites have been fully identified in excreta
and 8 metabolites in plasma. The major metabolite identified was O-desmethyl gefitinib, which is
14-fold less potent than gefitinib at inhibiting EGFR stimulated cell growth and has no inhibitory
effect on tumour cell growth in mice. It is therefore considered unlikely that it contributes to the
clinical activity of gefitinib.
The formation of O-desmethyl gefitinib has been shown,
in vitro
, to be via CYP2D6. The role of
CYP2D6 in the metabolic clearance of gefitinib has been evaluated in a clinical trial in healthy
volunteers genotyped for CYP2D6 status. In poor metabolisers no measurable levels of O-desmethyl
gefitinib were produced. The levels of exposure to gefitinib achieved in both the extensive and the
poor metaboliser groups were wide and overlapping but the mean exposure to gefitinib was 2-fold
higher in the poor metaboliser group. The higher average exposures that could be achieved by
13
Gender


individuals with no active CYP2D6 may be clinically relevant since adverse effects are related to dose
and exposure.
Elimination
Gefitinib is excreted mainly as metabolites via the faeces, with renal elimination of gefitinib and
metabolites accounting for less than 4 % of the administered dose.
Gefitinib total plasma clearance is approximately 500 ml/min and the mean terminal half-life is 41
hours in cancer patients. Administration of gefitinib once daily results in 2 to 8-fold accumulation,
with steady state exposures achieved after 7 to 10 doses. At steady state, circulating plasma
concentrations are typically maintained within a 2- to 3-fold range over the 24-hour dosing interval.
Special populations
From analyses of population pharmacokinetic data in cancer patients, no relationships were identified
between predicted steady state trough concentration and patient age, body weight, gender, ethnicity or
creatinine clearance (above 20 ml/min).
Hepatic impairment
In a phase I open-label study of single dose gefitinib 250 mg in patients with mild, moderate or severe
hepatic impairment due to cirrhosis (according to Child-Pugh classification), there was an increase in
exposure in all groups compared with healthy controls. An average 3.1-fold increase in exposure to
gefitinib in patients with moderate and severe hepatic impairment was observed. None of the patients
had cancer, all had cirrhosis and some had hepatitis. This increase in exposure may be of clinical
relevance since adverse experiences are related to dose and exposure to gefitinib.
Gefitinib has been evaluated in a clinical trial conducted in 41 patients with solid tumours and normal
hepatic function, or moderate or severe hepatic impairment (classified according to baseline Common
Toxicity Criteria grades for AST, alkaline phosphatase and bilirubin) due to liver metastases. It was
shown that following daily administration of 250 mg gefitinib, time to steady state, total plasma
clearance (C
maxSS)
and steady-state exposure (AUC
24SS
) were similar for the groups with normal and
moderately impaired hepatic function. Data from 4 patients with severe hepatic impairment due to
liver metastases suggested that steady-state exposures in these patients are also similar to those in
patients with normal hepatic function.
5.3 Preclinical safety data
Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to the
clinical exposure levels and with possible relevance to clinical use were as follows:
− Corneal epithelia atrophy and corneal translucencies
− Renal papillary necrosis
− Hepatocellular necrosis and eosinophilic sinusoidal macrophage infiltration
Data from
in vitro
studies indicate that gefitinib has the potential to inhibit cardiac repolarization (e.g.
QT interval). The clinical significance of these findings is unknown.
A reduction in female fertility was observed in the rat at a dose of 20 mg/kg/day.
Published studies have shown that genetically modified mice, lacking expression of EGFR, exhibit
developmental defects, related to epithelial immaturity in a variety of organs including the skin,
gastrointestinal tract and lung. When gefitinib was administered to rats during organogenesis, there
were no effects on embryofoetal development at the highest dose (30 mg/kg/day). However, in the
rabbit, there were reduced foetal weights at 20 mg/kg/day and above. There were no
compound-induced malformations in either species. When administered to the rat throughout gestation
and parturition, there was a reduction in pup survival at a dose of 20 mg/kg/day.
14
Following oral administration of C-14 labelled gefitinib to lactating rats 14 days post partum,
concentrations of radioactivity in milk were 11-19 fold higher than in blood.
Gefitinib showed no genotoxic potential.
A 2-year carcinogenicity study in rats resulted in a small but statistically significant increased
incidence of hepatocellular adenomas in both male and female rats and mesenteric lymph node
haemangiosarcomas in female rats at the highest dose (10 mg/kg/day) only. The hepatocellular
adenomas were also seen in a 2-year carcinogenicity study in mice, which demonstrated a small
increased incidence of this finding in male mice at the mid dose, and in both male and female mice at
the highest dose. The effects reached statistical significance for the female mice, but not for the males.
At no-effect levels in both mice and rats there was no margin in clinical exposure. The clinical
relevance of these findings is unknown.
The results of an
in vitro
phototoxicity study demonstrated that gefitinib may have phototoxicity
potential.
6.
6.1 List of excipients
Tablet core:
Lactose monohydrate
Microcrystalline cellulose (E460)
Croscarmellose sodium
Povidone (K29-32) (E1201)
Sodium laurilsulfate
Magnesium stearate
Tablet coating:
Hypromellose (E464)
Macrogol 300
Titanium dioxide (E171)
Yellow iron oxide (E172)
Red iron oxide (E172)
6.2 Incompatibilities
Not applicable.
6.3 Shelf life
4 years.
6.4 Special precautions for storage
Store in the original package in order to protect from moisture.
6.5 Nature and contents of container
PVC/Aluminium perforated blister containing 10 tablets or PVC/Aluminium non-perforated blister
containing 10 tablets.
3 blisters are combined with an aluminium foil laminate over-wrap in a carton.
Pack size of 30 film-coated tablets.
Not all pack sizes may be marketed.
15
6.6 Special precautions for disposal
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
AstraZeneca AB
S-151 85
Sodertalje
Sweden
8.
EU/1/09/526/001
EU/1/09/526/002
9.
24/06/2009
Detailed information on this product is available on the website of the European Medicines Agency
http://www.ema.europa.eu.
16
ANNEX II
A. MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
B. CONDITIONS OF THE MARKETING AUTHORISATION
17
A. MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturer responsible for batch release
AstraZeneca UK Limited
Macclesfield
Cheshire SK10 2NA
United Kingdom
B. CONDITIONS OF THE MARKETING AUTHORISATION
Medicinal product subject to restricted medical prescription (See Annex I: Summary of Product
Characteristics, section 4.2).
•
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND
EFFECTIVE USE OF THE MEDICINAL PRODUCT
Not applicable.
•
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, as described in version 10.0 presented in
Module 1.8.1. of the Marketing Authorisation Application, is in place and functioning before and
whilst the product is on the market.
Risk Management Plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in
the Pharmacovigilance Plan, as agreed in version 6 of the Risk Management Plan (RMP) presented in
Module 1.8.2. of the Marketing Authorisation Application and any subsequent updates of the RMP
agreed by the CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, the
updated RMP should be submitted at the same time as the next Periodic Safety Update Report
(PSUR).
In addition, an updated RMP should be submitted
• When new information is received that may impact on the current Safety Specification,
Pharmacovigilance Plan or risk minimisation activities
• Within 60 days of an important (pharmacovigilance or risk minimisation) milestone being
reached
• At the request of the European Medicines Agency.
18
ANNEX III
LABELLING AND PACKAGE LEAFLET
19
A. LABELLING
20
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
CARTON
1.
IRESSA 250 mg film-coated tablets
gefitinib
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each tablet contains 250 mg gefitinib.
3.
LIST OF EXCIPIENTS
Contains lactose monohydrate, see package leaflet for further information.
4.
30 film-coated tablets.
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Read the package leaflet before use.
Oral use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
9.
SPECIAL STORAGE CONDITIONS
Store in the original package in order to protect from moisture.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
21
























AstraZeneca AB
S-151 85
Sodertalje
Sweden
EU/1/09/526/001
EU/1/09/526/002
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
IRESSA
22

















MINIMUM PARTICULARS TO APPEAR ON BLISTERS OR STRIPS
BLISTER/ALUMINUM FOIL LAMINATE FLOW WRAP
1.
IRESSA 250 mg tablets
gefitinib
2.
AstraZeneca
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
23













B. PACKAGE LEAFLET
24
PACKAGE LEAFLET: INFORMATION FOR THE USER
IRESSA 250
mg film-coated tablets
gefitinib
Read all of this leaflet carefully before you start taking this medicine.
− Keep this leaflet. You may need to read it again.
− If you have any further questions, ask your doctor or pharmacist.
− This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even if
their symptoms are the same as yours.
− If any of the side effects get serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor or pharmacist.
In this leaflet
:
1.
What IRESSA is and what it is used for
2.
Before you take IRESSA
4.
Possible side effects
5.
How to store IRESSA
6.
Further information
1.
WHAT IRESSA IS AND WHAT IT IS USED FOR
IRESSA contains the active substance gefitinib which blocks a protein called ‘epidermal growth factor
receptor’ (EGFR). This protein is involved in the growth and spread of cancer cells.
IRESSA is used to treat adults with non-small cell lung cancer. This cancer is a disease in which
malignant (cancer) cells form in the tissues of the lung.
2.
BEFORE YOU TAKE IRESSA
Do not take IRESSA
− if you are allergic (hypersensitive) to gefitinib or any of the other ingredients of IRESSA (listed in
section 6, ‘What IRESSA contains’).
− if you are breast-feeding.
Take special care with IRESSA
Check with your doctor or pharmacist before taking IRESSA
− if you have ever had any other lung problems. Some lung problems may get worse during
treatment with IRESSA.
− if you have ever had problems with your liver.
Using other medicines
Please tell your doctor or pharmacist if you are taking or have recently taken any other medicines,
including medicines obtained without a prescription and herbal medicines.
In particular, tell your doctor or pharmacist if you are taking any of the following medicines:
− Phenytoin or carbamazepine (for epilepsy).
− Rifampicin (for tuberculosis).
− Itraconazole (for fungal infections).
− Barbiturates (a type of medicine used for sleeping problems).
25
3.
How to take IRESSA
− Herbal remedies containing St John’s wort (
Hypericum perforatum
, used for depression and
anxiety).
− Proton-pump inhibitors, H
2
-antagonists and antacids (for ulcers, indigestion, heartburn and to
reduce acids in the stomach).
These medicines may affect the way IRESSA works.
− Warfarin (a so-called oral anticoagulant, to prevent blood clots). If you are taking a medicine
containing this active substance, your doctor may need to do blood tests more often.
If any of the above applies to you, or if you are not sure, check with your doctor or pharmacist before
taking IRESSA.
Pregnancy and breast-feeding
Talk to your doctor before taking this medicine if you are pregnant, may become pregnant or are
breast-feeding.
It is recommended that you avoid becoming pregnant during treatment with IRESSA because IRESSA
could harm your baby.
Do not take IRESSA if you are breast-feeding.
Driving and using machines
IRESSA has no or negligible influence on your ability to drive or use any tools or machines.
However, if you feel weak whilst taking this medicine, take care driving or using tools or machines.
Important information about some of the ingredients of IRESSA
This medicine contains lactose. If you have been told by your doctor that you have an intolerance to
some sugars, contact your doctor before taking this medicine.
3.
HOW TO TAKE IRESSA
Always take IRESSA exactly as your doctor has told you. You should check with your doctor or
pharmacist if you are not sure.
• The usual dose is one 250 mg tablet per day.
• Take the tablet at about the same time each day.
• You can take the tablet with or without food.
• Do not take antacids (to reduce the acid level of your stomach) 2 hours before or 1 hour after
taking IRESSA.
If you have trouble swallowing the tablet, dissolve it in half a glass of still (non-fizzy) water. Do not
use any other liquids. Do not crush the tablet. Swirl the water until the tablet has dissolved. This may
take up to 20 minutes. Drink the liquid straight away. To make sure that you have drunk all of the
medicine, rinse the glass very well with half a glass of water and drink it.
If you take more IRESSA than you should
If you have taken more tablets than you should, talk to a doctor or pharmacist straight away.
If you forget to take IRESSA
What to do if you forget to take a tablet, depends on how long it is until your next dose.
• If it is 12 hours or more until your next dose:
take the missed tablet as soon as you remember.
Then take the next dose as usual.
• If it is less than 12 hours until your next dose:
skip the missed tablet. Then take the next tablet
at the usual time.
Do not take a double dose (two tablets at the same time) to make up for a forgotten dose.
If you have any further questions on the use of this product, ask your doctor or pharmacist.
4.
POSSIBLE SIDE EFFECTS
26
Like all medicines, IRESSA can cause side effects, although not everybody gets them.
These side effects may occur with certain frequencies, which are defined as follows:
• very common: affects more than 1 user in 10
• common: affects 1 to 10 users in 100
• uncommon: affects 1 to 10 users in 1,000
• rare: affects 1 to 10 users in 10,000
• very rare: affects less than 1 user in 10,000
• not known: frequency cannot be estimated from the available data.
Tell your doctor immediately if you notice any of the following side effects - you may need
urgent medical treatment:
• Allergic reaction (uncommon), particularly if symptoms include
swollen face, tongue or
throat, difficulty to swallow, hives and difficulties to breathe.
• Serious breathlessness, or sudden worsening breathlessness, possibly with a cough or fever.
This may mean that you have an inflammation of the lungs called ‘interstitial lung disease’.
This may affect about 1 in 100 patients taking IRESSA and can be life-threatening.
• Severe skin reactions (rare) affecting large areas of your body. The signs may include redness,
pain, ulcers, blisters, and shedding of the skin. The lips, nose, eyes and genitals may also be
affected.
• Dehydration (common) caused by long term or severe diarrhoea, vomiting (being sick),
nausea (feeling sick) or loss of appetite.
• Eye problems (uncommon), such as pain, redness changes in vision or ingrowing eyelashes.
This may mean that you have an ulcer on the surface of the eye (cornea).
Tell your doctor if you notice any of the following side effects:
Very common
side effects
• Diarrhoea
• Vomiting
• Nausea
• Skin reactions such as an acne-like rash, which is sometimes itchy with dry and/or cracked
skin
• Loss of appetite
• Weakness
• Dry, red or sore mouth
• Increase of a liver enzyme known as alanine aminotransferase in a blood test; if too high, your
doctor may tell you to stop taking IRESSA
Common side effects
• Dry, red or itchy eyes
• Red and sore eyelids
• Nail problems
• Hair loss
• Fever
• Bleeding (such as nose bleed or blood in your urine)
• Protein in your urine (shown in a urine test)
• Increase of bilirubin and the other liver enzyme known as aspartate aminotransferase in a
blood test; if too high, your doctor may tell you to stop taking IRESSA
• Increase of creatinine levels in a blood test (related to kidney function).
• cystitis (b
urning sensations during urination and frequent, urgent need to urinate
)
Uncommon
side effects
27
• Inflammation of the pancreas. The signs include very severe pain in the upper part of the
stomach area and severe nausea and vomiting.
• Inflammation of the liver. Symptoms may include a general feeling of being unwell, with or
without possible jaundice (yellowing of the skin and eyes). This side effect is uncommon;
however, some patients have died from this.
• Gastrointestinal perforation
Rare side effects
• Inflammation of the blood vessels in the skin. This may give the appearance of bruising or
patches of non-blanching rash on the skin.
• Haemorrhagic cystitis (
burning sensations during urination and frequent, urgent need to
urinate
with blood in the urine)
If any of the side effects get serious, or if you notice any side effects not listed in this leaflet, please
tell your doctor or pharmacist.
5.
HOW TO STORE IRESSA
Keep out of the reach and sight of children.
Do not use IRESSA after the expiry date which is stated on the carton, blister and overwrap foil after
EXP. The expiry date refers to the last day of that month.
Store in the original package in order to protect from moisture.
Medicines should not be disposed of via wastewater or household waste. Ask your pharmacist how to
dispose of medicines no longer required. These measures will help to protect the environment.
6.
FURTHER INFORMATION
What IRESSA contains
• The active substance is gefitinib. Each tablet contains 250 mg of gefitinib.
• The other ingredients are lactose monohydrate, microcrystalline cellulose (E460), croscarmellose
sodium, povidone (K29-32) (E1201), sodium laurilsulfate, magnesium stearate, hypromellose
(E464), macrogol 300, titanium dioxide (E171), yellow iron oxide (E172) and red iron oxide
(E172).
What IRESSA looks like and contents of the pack
IRESSA is a round brown tablet marked with ‘IRESSA 250’ on one side and plain on the other.
IRESSA comes in blister packs of 30 tablets. The blister foil may be perforated or non-perforated.
Marketing Authorisation Holder
AstraZeneca AB
S-151 85 Sodertalje
Sweden
Manufacturer
AstraZeneca UK Limited
Macclesfield
Cheshire SK10 2NA
United Kingdom
28
For any information about this medicine, please contact the local representative of the Marketing
Authorisation Holder (see contacts list):
België/Belgique/Belgien
NV AstraZeneca SA
Tel: +32 2 370 48 11
Luxembourg/Luxemburg
NV AstraZeneca SA
Tél/Tel: +32 2 370 48 11
България
ТП AstraZeneca UK Limited
Тел.: +359 2 971 25 33
Magyarország
AstraZeneca kft
Tel.: +36 1 883 6500
Česká republika
AstraZeneca Czech Republic s.r.o.
Tel: +420 222 807 111
Malta
Associated Drug Co. Ltd
Tel: +356 2277 8000
Danmark
AstraZeneca A/S
Tlf: +45 43 66 64 62
Nederland
AstraZeneca BV
Tel: +31 79 363 2222
Deutschland
AstraZeneca GmbH
Tel: +49 41 03 7080
Norge
AstraZeneca AS
Tlf: +47 21 00 64 00
Eesti
AstraZeneca
Tel: +372 6549 600
Österreich
AstraZeneca Österreich GmbH
Tel: +43 1 711 31 0
Ελλάδα
AstraZeneca A.E.
Τηλ: +30 2 106871500
Polska
AstraZeneca Pharma Poland Sp. z o.o.
Tel.: +48 22 874 35 00
España
AstraZeneca Farmacéutica Spain, S.A.
Tel: +34 91 301 91 00
Portugal
AstraZeneca Produtos Farmacêuticos, Lda.
Tel: +351 21 434 61 00
France
AstraZeneca
Tél: +33 1 41 29 40 00
România
AstraZeneca Pharma SRL
Tel: +40 21 317 60 41
Ireland
AstraZeneca Pharmaceuticals (Ireland) Ltd
Tel: +353 1609 7100
Slovenija
AstraZeneca UK Limited
Tel: +386 1 51 35 600
Ísland
Vistor hf.
Sími: +354 535 7000
Slovenská republika
AstraZeneca AB o.z.
Tel: +421 2 5737 7777
Italia
AstraZeneca S.p.A.
Tel: +39 02 98011
Suomi/Finland
AstraZeneca Oy
Puh/Tel: +358 10 23 010
Κύπρος
Αλέκτωρ Φαρµακευτική Λτδ
Sverige
AstraZeneca AB
29
Τηλ: +357 22490305
Tel: +46 8 553 26 000
Latvija
AstraZeneca AB pārstāvniecība Latvijā
Tel: +371 67377100
United Kingdom
AstraZeneca UK Ltd
Tel: +44 1582 836 836
Lietuva
UAB AstraZeneca
Lietuva
Tel: +370 5 2660550
This leaflet was last approved in
{MM/YYYY}
Detailed information on this medicine is available on the European Medicines Agency web site:
http://www.ema.europa.eu
.
30
Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/iressa.html
Copyright © 1995-2021 ITA all rights reserved.