










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Lucentis 10 mg/ml solution for injection
2.
One ml contains 10 mg ranibizumab. Each vial contains 2.3 mg of ranibizumab in 0.23 ml solution.
Ranibizumab is a humanised monoclonal antibody fragment produced in
Escherichia coli
cells by
recombinant DNA technology.
For a full list of excipients, see section 6.1.
3.
Solution for injection
Sterile, clear, colourless to pale yellow aqueous solution.
4.
Lucentis is indicated in adults for:
the treatment of neovascular (wet) age-related macular degeneration (AMD) (see section 5.1).
the treatment of visual impairment due to diabetic macular oedema (DME) (see section 5.1).
Single-use vial for intravitreal use only.
Lucentis must be administered by a qualified ophthalmologist experienced in intravitreal injections.
Treatment of wet AMD
In wet AMD, the recommended dose for Lucentis is 0.5 mg given monthly as a single intravitreal
injection. This corresponds to an injection volume of 0.05 ml.
Lucentis treatment is initiated with a loading phase of one injection per month for three consecutive
months, followed by a maintenance phase in which patients should be monitored for visual acuity on a
monthly basis. If the patient experiences a loss of greater than 5 letters in visual acuity (ETDRS or
one Snellen line equivalent), Lucentis should be administered. The interval between two doses should
not be shorter than 1 month.
Treatment of visual impairment due to DME
In visual impairment due to DME, the recommended dose for Lucentis is 0.5 mg given as a single
intravitreal injection. This corresponds to an injection volume of 0.05 ml.
Treatment is given monthly and continued until maximum visual acuity is achieved i.e the patient‟s
visual acuity is stable for three consecutive monthly assessments performed while on ranibizumab
treatment. Consequently, if there is no improvement in visual acuity over the course of three
injections, continued treatment is not recommended.
Thereafter patients should be monitored monthly for visual acuity.
2
Treatment is resumed when monitoring indicates loss of visual acuity due to
DME. Monthly
injections should then be administered until stable visual acuity is reached again for three consecutive
monthly assessments (implying a minimum of two injections). The interval between two doses should
not be shorter than 1 month.
Lucentis and laser photocoagulation in DME
There is some experience of Lucentis administered concomitantly with laser photocoagulation (see
section 5.1). When given on the same day, Lucentis should be administered at least 30 minutes after
laser photocoagulation. Lucentis can be administered in patients who have received previous laser
photocoagulation.
Method of administration
As with all medicinal products for parenteral use, Lucentis should be inspected visually for particulate
matter and discoloration prior to administration.
Before treatment, the patient should be instructed to self-administer antimicrobial drops (four times
daily for 3 days before and following each injection).
The injection procedure should be carried out under aseptic conditions, which includes the use of
surgical hand disinfection, sterile gloves, a sterile drape and a sterile eyelid speculum (or equivalent)
and the availability of sterile paracentesis (if required). The patient‟s medical history for
hypersensitivity reactions should be carefully evaluated prior to performing the intravitreal procedure
(see section 4.4). The periocular skin, eyelid and ocular surface should be disinfected and adequate
anaesthesia and a broad-spectrum topical microbicide should be administered prior to the injection.
For information on preparation of Lucentis, see section 6.6.
The injection needle should be inserted 3.5-4.0 mm posterior to the limbus into the vitreous cavity,
avoiding the horizontal meridian and aiming towards the centre of the globe. The injection volume of
0.05 ml is then delivered; a different scleral site should be used for subsequent injections.
Additional information on special populations
Hepatic impairment
Lucentis has not been studied in patients with hepatic impairment. However, no special considerations
are needed in this population.
Renal impairment
Dose adjustment is not needed in patients with renal impairment (see section 5.2).
Paediatric population
Lucentis is not recommended for use in children and adolescents due to a lack of data on safety and
efficacy in these sub-populations.
Elderly
No dose adjustment is required in the elderly. There is limited experience in patients older than
75 years with DME.
Ethnicity
Experience with treatment is limited in groups other than Caucasians.
3
Hypersensitivity to the active substance or to any of the excipients.
Patients with active or suspected ocular or periocular infections.
Patients with active severe intraocular inflammation.
Treatment with Lucentis is for intravitreal injection only.
Intravitreous injections, including those with Lucentis, have been associated with endophthalmitis,
intraocular inflammation, rhegmatogenous retinal detachment, retinal tear and iatrogenic traumatic
cataract (see section 4.8). Proper aseptic injection techniques must always be used when
administering Lucentis. In addition, patients should be monitored during the week following the
injection to permit early treatment if an infection occurs. Patients should be instructed to report any
symptoms suggestive of endophthalmitis or any of the above mentioned events without delay.
Increases in intraocular pressure have been seen within 60 minutes of injection of Lucentis (see
section 4.8). Both intraocular pressure and the perfusion of the optic nerve head must therefore be
monitored and managed appropriately.
The safety and efficacy of Lucentis therapy administered to both eyes concurrently have not been
studied. If bilateral treatment is performed at the same time this could lead to an increased systemic
exposure, which could increase the risk of systemic adverse events.
As with all therapeutic proteins, there is a potential for immunogenicity with Lucentis. Since there is a
potential for an increased systemic exposure in subjects with DME, an increased risk for developing
hypersensitivity in this patient population cannot be excluded. Patients should also be instructed to
report if an intraocular inflammation increases in severity, which may be a clinical sign attributable to
intraocular antibody formation.
Lucentis should not be administered concurrently with other anti-VEGF (vascular endothelial growth
factor) agents (systemic or ocular).
The dose should be withheld and treatment should not be resumed earlier than the next scheduled
treatment in the event of:
a decrease in best-corrected visual acuity (BCVA) of ≥30 letters compared with the last
assessment of visual acuity;
an intraocular pressure of ≥30 mmHg;
a retinal break;
a subretinal haemorrhage involving the centre of the fovea, or, if the size of the haemorrhage is
≥50%, of the total lesion area;
performed or planned intraocular surgery within the previous or next 28 days.
Risk factors associated with the development of a retinal pigment epithelial tear after anti-VEGF
therapy for wet AMD, include a large and/or high pigment epithelial retinal detachment. When
initiating Lucentis therapy, caution should be used in patients with these risk factors for retinal
pigment epithelial tears.
Treatment should be discontinued in subjects with rhegmatogenous retinal detachment or stage 3 or 4
macular holes.
There is only limited experience in the treatment of subjects with DME due to type I diabetes.
Lucentis has not been studied in patients who have previously received intravitreal injections, in
4
patients with active systemic infections, proliferative diabetic retinopathy, or in patients with
concurrent eye conditions such as retinal detachment or macular hole. There is also no experience of
treatment with Lucentis in diabetic patients with an HbA1c over 12% and uncontrolled hypertension.
There are no data on safety in the treatment of DME patients with prior history of stroke or transient
ischaemic attacks. Since there is
a potential risk of arterial thromboembolic events following
intravitreal use of VEGF (vascular endothelial growth factor) inhibitors caution should be exercised
when treating such patients (see section 4.8)
.
No formal interaction studies have been performed.
For the adjunctive use of verteporfin photodynamic therapy (PDT) and Lucentis in wet AMD, see
section 5.1.
For the adjunctive use of laser photocoagulation and Lucentis in DME, see sections 4.2 and 5.1.
Women of childbearing potential/contraception in females
Women of childbearing potential should use effective contraception during treatment.
Pregnancy
For ranibizumab no clinical data on exposed pregnancies are available. Studies in cynomolgus
monkeys do not indicate direct or indirect harmful effects with respect to pregnancy or
embryonal/foetal development (see section 5.3). The systemic exposure to ranibizumab is low after
ocular administration, but due to its mechanism of action, ranibizumab must be regarded as
potentially teratogenic and embryo-/foetotoxic. Therefore, ranibizumab should not be used during
pregnancy unless the expected benefit outweighs the potential risk to the foetus. For women who wish
to become pregnant and have been treated with ranibizumab, it is recommended to wait at least
3 months after the last dose of ranibizumab before conceiving a child.
Breast-feeding
It is unknown whether Lucentis is excreted in human milk. Breast-feeding is not recommended during
the use of Lucentis.
The Lucentis treatment procedure may induce temporary visual disturbances, which may affect the
ability to drive or use machines (see section 4.8). Patients who experience these signs must not drive
or use machines until these temporary visual disturbances subside.
Wet AMD population
In AMD a total of 1,315 patients constituted the safety population in the three phase III studies with
24 months exposure to Lucentis and 440 patients were treated with the recommended dose of 0.5 mg.
Serious adverse events related to the injection procedure included endophthalmitis, rhegmatogenous
retinal detachment, retinal tear and iatrogenic traumatic cataract (see section 4.4).
Other serious ocular events observed among Lucentis-treated patients included intraocular
inflammation and increased intraocular pressure (see section 4.4).
The adverse events listed below occurred at a higher rate (at least 2 percentage points) in patients
5
receiving treatment with Lucentis 0.5 mg than in those receiving control treatment (sham or
verteporfin PDT) in the three controlled wet AMD phase III studies FVF2598g (MARINA),
FVF2587g (ANCHOR) and FVF3192g (PIER). These were therefore considered potential adverse
drug reactions. The safety data described below also include all adverse events (in at least
0.5 percentage points of patients) suspected to be at least potentially related to the injection procedure
or medicinal product in the 440 patients of the combined 0.5 mg treatment groups in wet AMD.
The adverse events are listed by system organ class and frequency using the following convention:
very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000
to <1/1,000), very rare (<1/10,000), not known (cannot be estimated from the available data). Within
each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
DME population
The safety of Lucentis was studied in a one-year sham-controlled trial (RESOLVE) and in a one-year
laser-controlled trial (RESTORE) conducted respectively in 102 and 235 ranibizumab-treated patients
with visual impairment due to DME (see section 5.1). The event of urinary tract infection, in the
common frequency category, met the adverse reaction criteria for the table below; otherwise ocular
and non-ocular events in the RESOLVE and RESTORE trials were reported with a frequency and
severity similar to those seen in the wet AMD trials.
Infections and infestations
Very common
Nasopharyngitis
Common
Urinary tract infection*
Blood and lymphatic system disorders
Common
Anaemia
Immune system disorders
Common
Hypersensitivity
Psychiatric disorders
Common
Anxiety
Nervous system disorders
Very common
Headache
Eye disorders
Very common
Vitritis, vitreous detachment, retinal haemorrhage, visual
disturbance, eye pain, vitreous floaters, conjunctival
haemorrhage, eye irritation, foreign body sensation in eyes,
lacrimation increased, blepharitis, dry eye, ocular hyperaemia,
eye pruritus.
Common
Retinal degeneration, retinal disorder, retinal detachment,
retinal tear, detachment of the retinal pigment epithelium,
retinal pigment epithelium tear, visual acuity reduced, vitreous
haemorrhage, vitreous disorder, uveitis, iritis, iridocyclitis,
cataract, cataract subcapsular, posterior capsule opacification,
punctuate keratitis, corneal abrasion, anterior chamber flare,
vision blurred, injection site haemorrhage, eye haemorrhage,
conjunctivitis, conjunctivitis allergic, eye discharge, photopsia,
photophobia, ocular discomfort, eyelid oedema, eyelid pain,
conjunctival hyperaemia.
Uncommon
Blindness, endophthalmitis, hypopyon, hyphaema,
keratopathy, iris adhesion, corneal deposits, corneal oedema,
corneal striae, injection site pain, injection site irritation,
abnormal sensation in eye, eyelid irritation.
6
Respiratory, thoracic and mediastinal disorders
Common
Cough
Gastrointestinal disorders
Common
Nausea
Skin and subcutaneous tissue disorders
Common
Allergic reactions (rash, urticaria, pruritus, erythema)
Musculoskeletal and connective tissue disorders
Very common
Arthralgia
Investigations
Intraocular pressure increased
* observed only in DME population
Product-class-related adverse reactions
:
In the wet AMD phase III studies, the overall frequency of
non-ocular haemorrhages, an adverse event potentially related to systemic VEGF (vascular
endothelial growth factor) inhibition, was slightly increased in ranibizumab-treated patients.
However, there was no consistent pattern among the different haemorrhages. There is a theoretical
risk of arterial thromboembolic events following intravitreal use of VEGF inhibitors. A low incidence
rate of arterial thromboembolic events was observed in the Lucentis clinical trials in patients with
AMD and DME and there were no major differences between the groups treated with ranibizumab
compared to control.
Cases of accidental overdose have been reported from the clinical studies in wet AMD and post-
marketing data. Adverse reactions associated with these reported cases were intraocular pressure
increased, transient blindness, reduced visual acuity, corneal oedema, corneal pain, and eye pain. If an
overdose occurs, intraocular pressure should be monitored and treated, if deemed necessary by the
attending physician.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Antineovascularisation agents, ATC code: S01LA04
Ranibizumab is a humanised recombinant monoclonal antibody fragment targeted against human
vascular endothelial growth factor A (VEGF-A). It binds with high affinity to the VEGF-A isoforms
(e.g. VEGF
110
, VEGF
121
and VEGF
165
), thereby preventing binding of VEGF-A to its receptors
VEGFR-1 and VEGFR-2. Binding of VEGF-A to its receptors leads to endothelial cell proliferation
and neovascularisation, as well as vascular leakage, all of which are thought to contribute to the
progression of the neovascular form of age-related macular degeneration and diabetic macular oedema
causing visual impairment.
Treatment of wet AMD
In wet AMD, the clinical safety and efficacy of Lucentis have been assessed in three randomised,
double-masked, sham- or active-controlled studies of 24 months duration in patients with neovascular
AMD. A total of 1,323 patients (879 active and 444 control) were enrolled in these studies.
In study FVF2598g (MARINA), 716 patients with minimally classic or occult with no classic
7
Very common
choroidal neovascularisation (CNV) received monthly intravitreal injections of Lucentis 0.3 mg
(n=238) or 0.5 mg (n=240) or sham (n=238) injections.
In study FVF2587g (ANCHOR), 423 patients with predominantly classic CNV lesions received
either: 1) monthly intravitreal injections of Lucentis 0.3 mg and sham PDT (n=140); 2) monthly
intravitreal injections of Lucentis 0.5 mg and sham PDT (n=140); or 3) sham intravitreal injections
and active verteporfin PDT (n=143). Sham or active verteporfin PDT was given with the initial
Lucentis injection and every 3 months thereafter if fluorescein angiography showed persistence or
recurrence of vascular leakage.
In both studies, the primary efficacy endpoint was the proportion of patients who maintained vision,
defined as losing <15 letters of visual acuity at 12 months compared with baseline. Approximately
95% of Lucentis-treated patients maintained their visual acuity. 34-40% of Lucentis-treated patients
experienced a clinically significant improvement in vision, defined as gaining 15 or more letters at
12 months, see Tables 1 and 2.
Table 1
Outcomes at Month 12 and Month 24 in study FVF2598g (MARINA)
Outcome measure
Month
Sham
(n=238)
Lucentis 0.5 mg
(n=240)
Loss of <15 letters in visual
acuity (%)
a
(maintenance of vision)
Month 12
62%
95%
Month 24
53%
90%
Gain of ≥15 letters in visual
acuity (%)
a
Month 12
5%
34%
Month 24
4%
33%
Mean change in visual acuity
(letters) (SD)
a
Month 12
-10.5 (16.6)
+7.2 (14.4)
Month 24
-14.9 (18.7)
+6.6 (16.5)
a
p0.01
Table 2
Outcomes at Month 12 and Month 24 in study FVF2587g (ANCHOR)
Outcome measure
Month
Verteporfin PDT
(n=143)
Lucentis 0.5 mg
(n=140)
Loss of <15 letters in visual
acuity (%)
a
(maintenance of vision)
Month 12
64%
96%
Month 24
66%
90%
Gain of ≥15 letters in visual
acuity (%)
a
Month 12
6%
40%
Month 24
6%
41%
Mean change in visual acuity
(letters) (SD)
a
Month 12
-9.5 (16.4)
+11.3 (14.6)
Month 24
-9.8 (17.6)
+10.7 (16.5)
a
p<0.01
8























Figure 1 Mean change in visual acuity from baseline to Month 24 in study FVF2598g
(MARINA) and study FVF2587g (ANCHOR)
Study FVF2598g (MARINA)
15
10
5
+6.6
0
-5
+ 21.5
-10
-15
-14.9
0
2
4
6
8
10
12
14
16
18
20
22
24
Month
Study FVF2587g (ANCHOR)
15
+10.7
10
5
0
+20.5
-5
-10
-9.8
-15
0
2
4
6
8
10
12
14
16
18
20
22
24
Month
MARINA
ANCHOR
LUCENTIS 0.5 mg (n=240)
LUCENTIS 0.5 mg (n=140)
Sham (n=238)
Verteporfin PDT (n=143)
Results from both trials indicated that continued ranibizumab treatment may also be of benefit in
patients who lost ≥15 letters of best-corrected visual acuity (BCVA) in the first year of treatment.
The use of Lucentis beyond 24 months has not been studied.
Study FVF3192g (PIER) was a randomised, double-masked, sham-controlled study designed to assess
the safety and efficacy of Lucentis in 184 patients with all forms of neovascular AMD. Patients
received Lucentis 0.3 mg (n=60) or 0.5 mg (n=61) intravitreal injections or sham (n=63) injections
once a month for 3 consecutive doses, followed by a dose administered once every 3 months. From
Month 14 of the study, sham-treated patients were allowed to cross over to receive ranibizumab and
from Month 19, more frequent treatments were possible. Patients treated with Lucentis in PIER
received a mean of 10 total treatments.
The primary efficacy endpoint was mean change in visual acuity at 12 months compared with
9



































































































































































































































































































































baseline. After an initial increase in visual acuity (following monthly dosing), on average, patients‟
visual acuity declined with quarterly dosing, returning to baseline at Month 12 and this effect was
maintained in most ranibizumab-treated patients (82%) at Month 24. Data from a limited number of
subjects that crossed over to receive ranibizumab after more than a year of sham-treatment suggested
that early initiation of treatment may be associated with a better preservation of visual acuity.
Data from an open label study (PROTECT) in 32 patients followed for 9 months in which the safety
of same-day administration of verteporfin PDT and Lucentis 0.5 mg was evaluated showed that the
incidence of intraocular inflammation following the initial treatment was 6.3% (2 of 32 patients).
In both the MARINA and ANCHOR studies, the improvement in visual acuity seen with Lucentis
0.5 mg at 12 months was accompanied by patient-reported benefits as measured by the National Eye
Institute Visual Function Questionnaire (VFQ-25) scores, a pre-specified secondary efficacy endpoint.
The differences between Lucentis 0.5 mg and the two control groups were assessed with p-values
ranging from 0.009 to <0.0001.
Treatment of visual impairment due to DME
The efficacy and safety of Lucentis have been assessed in two randomised, double-masked, sham- or
active controlled studies of 12 months duration in patients with visual impairment due to diabetic
macular oedema. A total of 496 patients (336 active and 160 control) were enrolled in these studies,
the majority had type II diabetes, 28 ranibizumab-treated patients had type I diabetes.
In the phase II study D2201 (RESOLVE), 151 patients were treated with ranibizumab (6 mg/ml, n=51,
10 mg/ml, n=51) or sham (n=49) by monthly intravitreal injections until pre-defined treatment
stopping criteria were met. The initial ranibizumab dose (0.3 mg or 0.5 mg) could be doubled at any
time during the study after the first injection. Laser photocoagulation was allowed as rescue treatment
at any time in the study after Month 3 in both treatment arms. The study had two parts: an exploratory
part (the first 42 patients analysed at Month 6) and a confirmatory part (the remaining 109 patients
analysed at Month 12).
In the confirmatory part of the study (2/3 of patients), the primary efficacy endpoint, the mean average
change in BCVA from Month 1 to Month 12 compared to baseline, was shown to be statistically
superior in ranibizumab-treated patients as compared to sham treatment, a result also confirmed in the
overall study population, with a mean of 10 injections. The superiority in the treatment effect was also
shown by the key secondary BCVA endpoints: mean change in BCVA at Month 12, and proportion of
patients with BCVA gain of ≥10 letters and ≥15 letters at 12 months, see Table 3.
10
Table 3
Outcomes at Month 12 in study D2201 (RESOLVE) (overall study population)
Outcome measure
Ranibizumab pooled
(n=102)
Sham
(n=49)
Mean average change in BCVA
from Month 1 to Month 12
compared to baseline (letters) (SD)
+7.8 (7.72)
-0.1 (9.77)
p-value
<0.0001
Mean change in BCVA at
Month 12 (letters) (SD)
+10.3 (9.14)
-1.4 (14.16)
p-value
<0.0001
Gain of ≥10 letters in BCVA (% of
patients) at Month 12
60.8
18.4
p-value
<0.0001
Gain of ≥15 letters in BCVA (% of
patients) at Month 12
32.4
10.2
p-value
0.0043
In the phase III study D2301 (RESTORE), 345 patients with visual impairment due to macular
oedema were randomised to receive either intravitreal injection of ranibizumab 0.5 mg as
monotherapy and sham laser photocoagulation (n=116), combined ranibizumab 0.5 mg and laser
photocoagulation (n=118), or sham injection and laser photocoagulation (n=111). Treatment with
ranibizumab was started with monthly intravitreal injections and continued until visual acuity was
stable for at least three consecutive monthly assessments. The treatment was reinitiated when a
reduction in BCVA due to DME progression was observed. Laser photocoagulation was administered
at baseline on the same day, at least 30 minutes before injection of ranibizumab, and then as needed
based on ETDRS criteria.
The primary efficacy endpoint of mean average change in BCVA from Month 1 to Month 12
compared to baseline was shown to be superior in patients treated with ranibizumab monotherapy or
adjunctive to laser photocoagulation compared to laser control, without relevant efficacy differences
between ranibizumab monotherapy and ranibizumab adjunctive to laser, with a mean of 7 injections.
A clinically relevant improvement in BCVA gain of ≥10 letters and ≥15 letters was also observed, as
well as superiority in the mean BCVA change at Month 12, see Table 4 and Figure 2.
Table 4
Outcomes at Month 12 in study D2301 (RESTORE)
Outcome measure compared to baseline
Ranibizumab
0.5 mg
n=115
Ranibizumab
0.5 mg + Laser
n=118
Laser
n=110
Mean average change in BCVA from
Month 1 to Month 12 (SD)
6.1 (6.4)
5.9 (7.9)
0.8 (8.6)
p-value
<0.0001
<0.0001
Gain of ≥10 letters or BCVA 84 (% of
patients)
37.4
43.2
15.5
p-value
<0.0001
<0.0001
Gain of ≥15 letters or BCVA 84 (% of
patients)
22.6
22.9
8.2
p-value
0.0032
0.0021
11

























































































Figure 2 Mean change in visual acuity from baseline over time in study D2301 (RESTORE)
12
10
8
+ 6.8/+ 6.4
6
4
+ 6.2/+ 5.4*
2
0
+ 0.9
-2
-4
0
1
2
3
4
5
6
7
8
9 10 11 12
Month
Treatment group
Ranibizumab 0.5 mg (N = 115)
Ranibizumab 0.5 mg + Laser (N = 118)
Laser (N = 110)
BL=baseline; SE=standard error of mean
* Difference in least square means, p
0.0001/0.0004 based on two-sided stratified Cochran-Mantel-Haenszel
test
The effect was consistent in most subgroups. However, subjects with a fairly good baseline BCVA
(>73 letters) together with macular oedema with central retinal thickness of <300 m did not appear
to benefit from treatment with ranibizumab compared to laser photocoagulation.
The improvement in visual acuity seen with Lucentis 0.5 mg at 12 months was accompanied by
patient-reported benefits with regards to most vision-related functions including the overall composite
score and specifically regarding the subscales „general vision‟, „near activities‟ and „distance
activities‟ as measured by the National Eye Institute Visual Function Questionnaire (VFQ-25) scores,
a pre-specified secondary efficacy endpoint. For other subscales of this questionnaire no treatment
differences could be established
.
The difference between Lucentis 0.5 mg and the control group was
assessed with p-values of 0.0137 (ranibizumab mono) and 0.0041 (ranibizumab+laser) for the VFQ-
25 composite score.
In both studies, the improvement of vision was accompanied by a continuous decrease in the macular
oedema as measured by central retinal thickness (CRT).
No new ocular and non-ocular safety findings were observed in any of the above DME studies.
Paediatric population
Safety and efficacy of ranibizumab have not yet been studied in paediatric patients.
The European Medicine Agency has waived the obligation to submit the results of studies with
Lucentis in all subsets of the paediatric population in visual impairment due to diabetic macular
oedema. See section 4.2 for information on paediatric use.
12










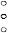







































































































































































5.2 Pharmacokinetic properties
Following monthly intravitreal administration of Lucentis to patients with neovascular AMD, serum
concentrations of ranibizumab were generally low, with maximum levels (C
max
) generally below the
ranibizumab concentration necessary to inhibit the biological activity of VEGF by 50% (11-27 ng/ml,
as assessed in an
in vitro
cellular proliferation assay). C
max
was dose proportional over the dose range
of 0.05 to 1.0 mg/eye. Serum concentrations in a limited number of DME patients indicate that a
slightly higher systemic exposure cannot be excluded compared to those observed in neovascular
AMD patients.
Based on analysis of population pharmacokinetics and disappearance of ranibizumab from serum for
patients with neovascular AMD treated with the 0.5 mg dose, the average vitreous elimination half-
life of ranibizumab is approximately 9 days. Upon monthly intravitreal administration of Lucentis
0.5 mg/eye, serum ranibizumab C
max
, attained approximately 1 day after dosing, is predicted to
generally range between 0.79 and 2.90 ng/ml, and C
min
is predicted to generally range between 0.07
and 0.49 ng/ml. Serum ranibizumab concentrations are predicted to be approximately 90,000-fold
lower than vitreal ranibizumab concentrations.
Patients with renal impairment: No formal studies have been conducted to examine the
pharmacokinetics of Lucentis in patients with renal impairment. Sixty-eight percent (136 of 200) of
patients in a population pharmacokinetic analysis had renal impairment (46.5% mild [50-80 ml/min],
20% moderate [30-50 ml/min], and 1.5% severe [<30 ml/min]). Systemic clearance was slightly
lower, but this was not clinically significant.
Hepatic impairment: No formal studies have been conducted to examine the pharmacokinetics of
Lucentis in patients with hepatic impairment.
5.3 Preclinical safety data
Bilateral intravitreal administration of ranibizumab to cynomolgus monkeys at doses between
0.25 mg/eye and 2.0 mg/eye once every 2 weeks for up to 26 weeks resulted in dose-dependent ocular
effects.
Intraocularly, there were dose-dependent increases in anterior chamber flare and cells with a peak
2 days after injection. The severity of the inflammatory response generally diminished with
subsequent injections or during recovery. In the posterior segment, there were vitreal cell infiltration
and floaters, which also tended to be dose-dependent and generally persisted to the end of the
treatment period. In the 26-week study, the severity of the vitreous inflammation increased with the
number of injections. However, evidence of reversibility was observed after recovery. The nature and
timing of the posterior segment inflammation is suggestive of an immune-mediated antibody response,
which may be clinically irrelevant. Cataract formation was observed in some animals after a relatively
long period of intense inflammation, suggesting that the lens changes were secondary to severe
inflammation. A transient increase in post-dose intraocular pressure was observed following
intravitreal injections, irrespective of dose.
Microscopic ocular changes were related to inflammation and did not indicate degenerative processes.
Granulomatous inflammatory changes were noted in the optic disc of some eyes. These posterior
segment changes diminished, and in some instances resolved, during the recovery period.
Following intravitreal administration, no signs of systemic toxicity were detected. Serum and vitreous
antibodies to ranibizumab were found in a subset of treated animals.
No carcinogenicity or mutagenicity data are available.
In pregnant monkeys, intravitreal ranibizumab treatment resulting in maximal systemic exposures
0.9-7-fold a worst case clinical exposure did not elicit developmental toxicity or teratogenicity, and
13
had no effect on weight or structure of the placenta, although, based on its pharmacological effect
ranibizumab should be regarded as potentially teratogenic and embryo-/foetotoxic.
The absence of ranibizumab-mediated effects on embryo-foetal development is plausibly related
mainly to the inability of the Fab fragment to cross the placenta. Nevertheless, a case was described
with high maternal ranibizumab serum levels and presence of ranibizumab in foetal serum, suggesting
that the anti-ranibizumab antibody acted as (Fc region containing) carrier protein for ranibizumab,
thereby decreasing its maternal serum clearance and enabling its placental transfer. As the embryo-
foetal development investigations were performed in healthy pregnant animals and disease (such as
diabetes) may modify the permeability of the placenta towards a Fab fragment, the study should be
interpreted with caution.
6.
6.1 List of excipients
α,α-trehalose dihydrate
Histidine hydrochloride, monohydrate
Histidine
Polysorbate 20
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other
medicinal products.
6.3 Shelf life
3 years
6.4 Special precautions for storage
Store in a refrigerator (2C - 8C).
Do not freeze.
Keep the vial in the outer carton in order to protect from light.
6.5 Nature and contents of container
0.23 ml solution in a vial (type I glass) with a stopper (chlorobutyl rubber), with 1 filter needle,
1 injection needle and 1 syringe (polypropylene). Pack containing 1 vial.
6.6 Special precautions for disposal and other handling
Vials are for single use only.
To prepare Lucentis for intravitreal administration, please adhere to the following instructions:
1.
Before withdrawal, the outer part of the rubber stopper of the vial should be disinfected.
2.
Assemble the 5 µm filter needle (provided) onto the 1 ml syringe (provided) using aseptic
technique. Push the blunt filter needle into the centre of the vial stopper until the needle touches
the bottom edge of the vial.
3.
Withdraw all the liquid from the vial, keeping the vial in an upright position, slightly inclined to
14
ease complete withdrawal.
4.
Ensure that the plunger rod is drawn sufficiently back when emptying the vial in order to
completely empty the filter needle.
5.
Leave the blunt filter needle in the vial and disconnect the syringe from the blunt filter needle.
The filter needle should be discarded after withdrawal of the vial contents and should not be
used for the intravitreal injection.
6.
Aseptically and firmly assemble the injection needle (provided) onto the syringe.
7.
Carefully remove the cap from the injection needle without disconnecting the injection needle
from the syringe.
Note: Grip at the yellow hub of the injection needle while removing the cap.
8.
Carefully expel the air from the syringe and adjust the dose to the 0.05 ml mark on the syringe.
The syringe is ready for injection.
Note: Do not wipe the injection needle. Do not pull back on the plunger.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
United Kingdom
8.
EU/1/06/374/001
9.
22.01.2007
Detailed information on this product is available on the website of the European Medicines Agency
http://www.ema.europa.eu
15
ANNEX II
A.
MANUFACTURER OF THE BIOLOGICAL ACTIVE
SUBSTANCE AND MANUFACTURING AUTHORISATION
HOLDER RESPONSIBLE FOR BATCH RELEASE
B.
CONDITIONS OF THE MARKETING AUTHORISATION
16
A. MANUFACTURER OF THE BIOLOGICAL ACTIVE SUBSTANCE AND
MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturer of the biological active substance
Genentech, Inc.
1 DNA Way
South San Francisco, CA 94080-4990
USA
Name and address of the manufacturer responsible for batch release
Novartis Pharma S.A.S.
Centre de Biotechnologie
8, rue de l'Industrie
F-68330 Huningue
France
B. CONDITIONS OF THE MARKETING AUTHORISATION
Medicinal product subject to restricted medical prescription. (See Annex I: Summary of Product
Characteristics, section 4.2)
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND
EFFECTIVE USE OF THE MEDICINAL PRODUCT
The
Marketing Authorisation Holder (
MAH) shall ensure all physicians who are expected to
prescribe/use Lucentis are provided with a physician information pack containing the following:
Physician information
Intravitreal injection procedure video
Intravitreal injection procedure pictogram
Patient information packs
The physician information should contain the following key elements:
The Summary of Product Characteristics
Sterile techniques, including periocular and ocular disinfection, to minimise risk of infection
Use of antibiotics
Use of povidone iodine or equivalent
Techniques for the intravitreal injection
Key signs and symptoms of IVT injection related adverse events
Management of IVT injection related adverse events
17
The patient information pack should be provided in the form of both patient information booklets and
audio-CD that contain following key elements:
Patient information leaflet
How to prepare for Lucentis treatment
What are the steps following treatment with Lucentis
Key signs and symptoms of serious adverse events
When to seek urgent attention from the health care provider
The MAH must implement this educational plan nationally, prior to marketing, and as agreed with the
competent authorities in the Member States.
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, as described in version 8.0 presented in
Module 1.8.1. of the Marketing Authorisation Application, is in place and functioning before and
whilst the product is on the market.
Risk Management Plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in
the Pharmacovigilance Plan, as agreed in version 7 dated 11 August 2010 of the Risk Management
Plan (RMP) presented in Module 1.8.2. of the Marketing Authorisation Application and any
subsequent updates of the RMP agreed by the CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, any
updated RMP should be submitted at the same time as the following Periodic Safety Update Report
(PSUR).
In addition, an updated RMP should be submitted:
When new information is received that may impact on the current Safety Specification,
Pharmacovigilance Plan or risk minimisation activities
Within 60 days of an important (pharmacovigilance or risk minimisation) milestone being
reached
At the request of the European Medicines Agency.
18
ANNEX III
LABELLING AND PACKAGE LEAFLET
19
A. LABELLING
20
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
CARTON
1.
Lucentis 10 mg/ml solution for injection
Ranibizumab
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
One ml contains 10 mg of ranibizumab. Vial containing 2.3 mg of ranibizumab.
3.
LIST OF EXCIPIENTS
Also contains: α,α-trehalose dihydrate; histidine hydrochloride, monohydrate; histidine;
polysorbate 20; water for injections.
4.
1 vial of 0.23 ml solution for injection
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Intravitreal use.
Vial for single use only.
Read the package leaflet before use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
21




















9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator.
Do not freeze.
Keep the vial in the outer carton in order to protect from light.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
United Kingdom
EU/1/06/374/001
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
-
22



















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
VIAL LABEL
1.
Lucentis 10 mg/ml solution for injection
Ranibizumab
Intravitreal use
2.
METHOD OF ADMINISTRATION
Read the package leaflet before use.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
1 vial = 2.3 mg ranibizumab.
6.
OTHER
23

















B. PACKAGE LEAFLET
24
PACKAGE LEAFLET: INFORMATION FOR THE USER
Lucentis 10 mg/ml solution for injection
Ranibizumab
Read all of this leaflet carefully before you are given this medicine.
-
Keep this leaflet. You may need to read it again.
-
If you have any further questions, ask your doctor.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
-
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor.
In this leaflet
:
1.
What Lucentis is and what it is used for
2.
Before you use Lucentis
3.
How to use Lucentis
4.
Possible side effects
5.
How to store Lucentis
1.
WHAT LUCENTIS IS AND WHAT IT IS USED FOR
Lucentis is given as an injection into the eye by your eye doctor under a local anaesthetic.
It is used to treat damage to the retina (the light-sensitive back part of the eye) when this damage is
caused by abnormal blood vessels growing and leaking into the eye which may decrease vision. This
happens in diseases such as wet age-related macular degeneration (AMD) or, in diabetic macular
oedema (DME). Lucentis can help to improve the damaged vision or stop it from getting worse.
The active substance in Lucentis is ranibizumab. Ranibizumab helps to stop the growth and leakage of
new blood vessels in the eye, abnormal processes that contribute to the progression of AMD and
DME.
2.
BEFORE YOU USE LUCENTIS
You must not receive Lucentis
-
if you are allergic (hypersensitive) to ranibizumab or any of the other ingredients of Lucentis
listed at the end of this leaflet (see section 6).
-
if you have an infection in or around your eye.
-
if you have pain or redness (severe intraocular inflammation) in your eye.
Take special care with Lucentis
-
Lucentis is given as an injection into the eye. Occasionally, an infection in the internal portion
of the eye, pain or redness (inflammation), detachment or tear of the layer in the back of the eye
(retinal detachment or tear), or clouding of the lens (cataract) may occur after Lucentis
treatment. It is important to identify and treat such an infection or retinal detachment as soon as
possible. Please tell your doctor immediately if you develop signs such as eye pain or increased
discomfort, worsening eye redness, blurred or decreased vision, an increased number of small
particles in your vision or increased sensitivity to light.
-
In some patients the eye pressure may increase for a short period directly after the injection.
This is something you may not notice, therefore your doctor may monitor this after each
injection.
25
6.
Further information
-
Inform your doctor if you have had a stroke or experienced transient signs of stroke (weakness
or paralysis of limbs or face, difficulty speaking or understanding). This information will be
taken into account to evaluate if Lucentis is the appropriate treatment for you.
Children and adolescents (below 18 years of age)
The use of Lucentis in children and adolescents has not been studied and is therefore not
recommended.
Using other medicines
Please tell your doctor if you are using or have recently used any other medicines, including
medicines bought without a prescription.
Pregnancy and breast-feeding
-
There is no experience of using Lucentis in pregnant women; therefore the potential risks are
unknown. If you are pregnant or planning to become pregnant, please discuss this with your
doctor before Lucentis treatment.
-
Lucentis is not recommended during breast-feeding because it is not known whether Lucentis
passes into human milk. Ask your doctor or pharmacist for advice before Lucentis treatment.
Driving and using machines
After Lucentis treatment you may experience some temporary vision blurring. If this happens, do not
drive or use machines until this resolves.
3.
HOW TO USE LUCENTIS
All Lucentis injections will be administered by your doctor.
Your doctor will ask you to use antimicrobial eye drops four times daily for 3 days before and after
each injection in order to prevent any possible eye infection.
Lucentis is administered as a single injection into your eye. The interval between two doses should
not be shorter than 1 month.
If you are being treated for wet age-related macular degeneration, the injection is given once a month
in the first 3 months. Afterwards, your doctor will monitor your vision on a monthly basis. If your
condition is found to be worsening, your doctor will administer Lucentis to your affected eye again.
If you are being treated for visual impairment due to diabetic macular oedema, the injection is given
once a month. Your doctor will monitor your vision monthly. If your vision remains the same while
given Lucentis treatment, your doctor may decide to stop the treatment with Lucentis. Your doctor
will continue to monitor your vision monthly and will decide if treatment with Lucentis should be
resumed or not. Your doctor may decide that you also need to be treated with laser for this condition.
If so, laser can be administered together with Lucentis.
Before the injection, your doctor will use antibiotic eye drops and wash your eye carefully to prevent
infection. Your doctor will also give you a local anaesthetic to reduce or prevent any pain you might
have with the injection.
Older people (age 65 years and over)
Lucentis can be used for people of 65 years of age and over without dose adjustment.
Use in children
Lucentis is not recommended for use in children and adolescents due to a lack of data on safety and
efficacy in this patient group.
26
If a dose of Lucentis is missed
Contact your doctor or hospital as soon as possible to re-schedule your appointment.
Before stopping Lucentis treatment
If you are considering stopping Lucentis treatment, please go to your next appointment and discuss
this with your doctor. Your doctor will advise you and decide how long you should be treated with
Lucentis.
If you have any further questions on the use of this product, ask your doctor.
4.
POSSIBLE SIDE EFFECTS
Like all medicines, Lucentis can cause side effects, although not everybody gets them. Please do not
be alarmed by this list of possible side effects. You may not experience any of them.
With administration of Lucentis, there may be some side effects, mostly in the eye and due to the
injection procedure. Occasionally an infection in the internal portion of the eye, detachment or tear of
the layer in the back of the eye (retinal detachment or tear), or clouding of the lens (cataract) may
occur in the two weeks after Lucentis treatment. Other side effects include pain or redness
(inflammation) and increased eye pressure. The symptoms you might experience are described in
section 2 of this leaflet (“Take special care with Lucentis”). Please read section 2. It tells you what to
do if you have any of these symptoms.
Very common side effects
(These may affect 10 or more in every 100 patients)
The most common side effects in the eye reported to be possibly caused by the medicinal product or
by the injection procedure include: Inflammation of the eye, blurred vision, bleeding in the back of
the eye (retinal bleeding), visual disturbances, eye pain, small particles or spots in your vision
(floaters), bloodshot eye, eye irritation, a feeling of having something in the eye, increased tear
production, inflammation or infection of the eyelid margins, dry eye, redness or itching of the eye.
Increased eye pressure has been observed very commonly.
The most common non-visual side effects reported to be possibly caused by the medicinal product or
by the injection procedure include: Sore throat, headache and joint pain.
Common side effects
(These may affect between 1 and 10 in every 100 patients)
Other common side effects in the eye reported to be possibly caused by the medicinal product or by
the injection procedure include: Seeing flashes of light with floaters progressing to a loss of sight,
decreased sharpness of vision, swelling of a section of the eye (uvea, cornea), clouding of the lens,
small marks on the surface of the eye, bleeding in the eye, discharge from the eye with itching,
redness and swelling (conjunctivitis), light sensitivity, eye discomfort, swelling of the eyelid, eyelid
pain.
Other common non-visual side effects reported to be possibly caused by the medicinal product or by
the injection procedure include: Urinary tract infection, fatigue, general feeling of being unwell,
anxiety, cough, nausea, allergic reactions like rash, itching, skin reddening.
Uncommon side effects
(These may affect less than 1 in every 100 patients)
Uncommon side effects in the eye reported to be possibly caused by the medicinal product or by the
injection procedure include: Blindness, infection of the eye globe (endophthalmitis), inflammation
and bleeding in the front part of the eye, sac of pus on the eye, changes of the central part of the eye
surface, pain or irritation at the site of injection, abnormal sensation in the eye, irritation of the eyelid.
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor.
27
5.
HOW TO STORE LUCENTIS
-
Keep out of the reach and sight of children.
-
Do not use Lucentis after the expiry date which is stated on the carton and vial label after EXP.
The expiry date refers to the last day of that month.
-
Store in a refrigerator (2°C – 8°C). Do not freeze.
-
Keep the vial in the outer carton in order to protect from light.
-
Do not use any pack that is damaged.
6.
FURTHER INFORMATION
What Lucentis contains
-
The active substance in Lucentis is ranibizumab (10 mg/ml). Each ml contains 10 mg
ranibizumab.
-
The other ingredients are: α,α-trehalose dihydrate; histidine hydrochloride, monohydrate;
histidine; polysorbate 20; water for injections.
What Lucentis looks like and contents of the pack
Lucentis is a solution for injection in a vial (0.23 ml). The solution is clear, colourless to pale yellow
and aqueous.
Lucentis is supplied as a pack containing one glass vial of ranibizumab with chlorobutyl rubber
stopper, one filter needle for withdrawal of the vial contents, one injection needle and one syringe for
withdrawal of the vial contents and for intravitreal injection.
Marketing Authorisation Holder
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
United Kingdom
Manufacturer
Novartis Pharma S.A.S.
Centre de Biotechnologie
8, rue de l‟Industrie
F-68330 Huningue
France
For any information about this medicine, please contact the local representative of the Marketing
Authorisation Holder:
België/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
България
Novartis Pharma Services Inc.
Тел.: +359 2 489 98 28
Magyarország
Novartis Hungária Kft. Pharma
Tel.: +36 1 457 65 00
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Malta
Novartis Pharma Services Inc.
Tel: +356 2298 3217
28
Danmark
Novartis Healthcare A/S
Tlf: +45 39 16 84 00
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Deutschland
Novartis Pharma GmbH
Tel: +49 911 273 0
Norge
Novartis Norge AS
Tlf: +47 23 05 20 00
Eesti
Novartis Pharma Services Inc.
Tel: +372 66 30 810
Österreich
Novartis Pharma GmbH
Tel: +43 1 86 6570
Ελλά
Novartis (Hellas) A.E.B.E.
Τηλ: +30 210 281 17 12
Polska
Novartis Poland Sp. z o.o.
Tel.: +48 22 550 8888
España
Novartis Farmacéutica, S.A.
Tel: +34 93 306 42 00
Portugal
Novartis Farma - Produtos Farmacêuticos, S.A.
Tel: +351 21 000 8600
France
Novartis Pharma S.A.S.
Tél: +33 1 55 47 66 00
România
Novartis Pharma Services Inc.
Tel: +40 21 31299 01
Ireland
Novartis Ireland Limited
Tel: +353 1 260 12 55
Slovenija
Novartis Pharma Services Inc.
Tel: +386 1 300 75 50
Ísland
Vistor hf.
Sími: +354 535 7000
Slovenská republika
Novartis Slovakia s.r.o.
Tel: +421 2 5542 5439
Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Suomi/Finland
Novartis Finland Oy
Puh/Tel: +358 (0)10 6133 200
Κύπρς
Novartis Pharma Services Inc.
Τηλ: +357 22 690 690
Sverige
Novartis Sverige AB
Tel: +46 8 732 32 00
Latvija
Novartis Pharma Services Inc.
Tel: +371 67 887 070
United Kingdom
Novartis Pharmaceuticals UK Ltd.
Tel: +44 1276 698370
Lietuva
Novartis Pharma Services Inc.
Tel: +370 5 269 16 50
This leaflet was last approved in
Detailed information on this medicine is available on the European Medicines Agency web site:
http://www.ema.europa.eu
29
INFORMATION FOR THE HEALTHCARE PROFESSIONAL
How to prepare and administer Lucentis
Single-use vial for intravitreal use only
Lucentis must be administered by a qualified ophthalmologist experienced in intravitreal injections.
In wet AMD and visual impairment due to DME, the recommended dose for Lucentis is 0.5 mg given
monthly as a single intravitreal injection. This corresponds to an injection volume of 0.05 ml.
In wet AMD, the treatment is initiated with a loading phase of one injection per month for three
consecutive months, followed by a maintenance phase in which patients should be monitored for
visual acuity on a monthly basis. If the patient experiences a loss of greater than 5 letters in visual
acuity (ETDRS or one Snellen line equivalent), Lucentis should be administered. The interval
between two doses should not be shorter than 1 month.
In visual impairment due to DME, treatment is given monthly and continued until maximum visual
acuity is achieved, confirmed by stable visual acuity for three consecutive monthly assessments
performed while on ranibizumab treatment. Thereafter patients should be monitored monthly for
visual acuity. Treatment is resumed with monthly injections when monitoring indicates loss of visual
acuity due to DME until stable visual acuity is reached again for three consecutive monthly
assessments. The interval between two doses should not be shorter than 1 month.
Lucentis and laser photocoagulation in DME
There is some experience of Lucentis administered concomitantly with laser photocoagulation. When
given on the same day, Lucentis should be administered at least 30 minutes after laser
photocoagulation. Lucentis can be administered in patients who have received previous laser
photocoagulation.
As with all medicinal products for parenteral use, Lucentis should be inspected visually for particulate
matter and discoloration prior to administration.
Before treatment, the patient should be instructed to self-administer antimicrobial drops (four times
daily for 3 days before and following each injection).
The injection procedure should be carried out under aseptic conditions, which includes the use of
surgical hand disinfection, sterile gloves, a sterile drape and a sterile eyelid speculum (or equivalent)
and the availability of sterile paracentesis (if required). The patient‟s medical history for
hypersensitivity reactions should be carefully evaluated prior to performing the intravitreal procedure.
The periocular skin, eyelid and ocular surface should be disinfected and adequate anaesthesia and a
broad-spectrum topical microbicide should be administered prior to the injection.
30
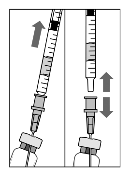
To prepare Lucentis for intravitreal administration, please adhere to the following instructions:
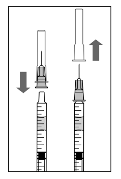
1. Before withdrawal, the outer part of the rubber stopper
of the vial should be disinfected.
2. Assemble the 5 µm filter needle (provided) onto the 1 ml
syringe (provided) using aseptic technique. Push the blunt
filter needle into the centre of the vial stopper until the
needle touches the bottom edge of the vial.
3. Withdraw all the liquid from the vial, keeping the vial in
an upright position, slightly inclined to ease complete
withdrawal.
4. Ensure that the plunger rod is drawn sufficiently back
when emptying the vial in order to completely empty the
filter needle.
5. Leave the blunt filter needle in the vial and disconnect
the syringe from the blunt filter needle. The filter needle
should be discarded after withdrawal of the vial contents
and should not be used for the intravitreal injection.
6. Aseptically and firmly assemble the injection needle
(provided) onto the syringe.
7. Carefully remove the cap from the injection needle
without disconnecting the injection needle from the
syringe.
Note: Grip at the yellow hub of the injection needle while
removing the cap.
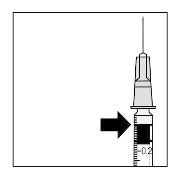
8. Carefully expel the air from the syringe and adjust the
dose to the 0.05 ml mark on the syringe. The syringe is
ready for injection.
Note: Do not wipe the injection needle. Do not pull back on
the plunger.
0.05 ml
The injection needle should be inserted 3.5-4.0 mm posterior to the limbus into the vitreous cavity,
avoiding the horizontal meridian and aiming towards the centre of the globe. The injection volume of
0.05 ml is then delivered; a different scleral site should be used for subsequent injections.
31

Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/lucentis.html
Copyright © 1995-2021 ITA all rights reserved.