










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Myozyme 50 mg powder for concentrate for solution for infusion
2.
One vial contains 50 mg of alglucosidase alfa.
After reconstitution, the solution contains 5 mg of alglucosidase* alfa per ml and after dilution, the
concentration varies from 0.5 mg to 4 mg/ml.
*Human acid α-glucosidase is produced in Chinese hamster ovary cells (CHO) by recombinant DNA
technology.
For a full list of excipients, see section 6.1.
3.
Powder for concentrate for solution for infusion.
White to off-white powder.
4.
Myozyme is indicated for long-term enzyme replacement therapy (ERT) in patients with a confirmed
diagnosis of Pompe disease (acid
term enzyme replacement therapy (ERT) in patients with a confirmed
-glucosidase deficiency).
Myozyme is indicated in adults and paediatric patients of all ages.
In patients with late-onset Pompe disease the evidence of efficacy is limited (see section 5.1).
Myozyme treatment should be supervised by a physician experienced in the management of patients
with Pompe disease or other inherited metabolic or neuromuscular diseases.
Posology
The recommended dose regimen of alglucosidase alfa is 20 mg/kg of body weight administered once
every 2 weeks.
Patient response to treatment should be routinely evaluated based on a comprehensive evaluation of
all clinical manifestations of the disease.
Paediatric and elderly
population
There is no evidence for special considerations when Myozyme is administered to paediatric patients
of all ages or elderly patients.
Renal and hepatic impairment
The safety and efficacy of Myozyme in patients with renal or hepatic impairment have not been
evaluated and no specific dose regimen can be recommended for these patients.
2
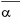
Method of administration
Myozyme should be administered as an intravenous infusion.
Infusions should be administered incrementally. It is recommended that the infusion begin at an initial
rate of 1 mg/kg/h and be gradually increased by 2 mg/kg/h every 30 minutes if there are no signs of
infusion associated reactions (IARs) until a maximum rate of 7 mg/kg/h is reached. IARs are
described in section 4.8.
For instructions on reconstitution and dilution of the medicinal product before administration, see
section 6.6.
Life threatening hypersensitivity (anaphylactic reaction) to the active substance or to any of the
excipients, when rechallenge was unsuccessful (see sections 4.4 and 4.8).
Hypersensitivity/Anaphylactic reactions
Serious and life-threatening anaphylactic reactions, including anaphylactic shock, have been reported
in infantile- and late-onset patients during Myozyme infusions (see section 4.8). Because of the
potential for severe infusion associated reactions, appropriate medical support measures, including
cardiopulmonary resuscitation equipment, should be readily available when Myozyme is
administered. If severe hypersensitivity or anaphylactic reactions occur, immediate discontinuation of
Myozyme infusion should be considered and appropriate medical treatment should be initiated. The
current medical standards for emergency treatment of anaphylactic reactions are to be observed.
Infusion Associated Reactions
Approximately half of the patients treated with Myozyme in infantile-onset clinical studies and 28%
of the patients treated with Myozyme in a late-onset clinical study developed infusion associated
reactions (IARs). IARs are defined as any related adverse event occurring during the infusion or
during the hours following infusion. Some reactions were severe (see section 4.8). A tendency was
observed in infantile patients treated with a higher dose (40 mg/kg) to experience more symptoms
when developing IARs. Infantile onset patients who develop high antibody titres appear to be at
higher risk for developing more frequent IARs. Patients with an acute illness (e.g. pneumonia, sepsis)
at the time of Myozyme infusion appear to be at greater risk for IARs. Careful consideration should be
given to the patient‟s clinical status prior to administration of Myozyme. Patients should be closely
monitored and all cases of IARs, delayed reactions and possible immunological reactions should be
reported to the marketing authorisation holder.
Patients who have experienced IARs (and in particular anaphylactic reactions) should be treated with
caution when re-administering Myozyme (see sections 4.3 and 4.8). Mild and transient effects may
not require medical treatment or discontinuation of the infusion. Reduction of the infusion rate,
temporary interruption of the infusion, or pre-treatment, generally with oral antihistamine and/or
antipyretics and/or corticosteroids, has effectively managed most reactions. IARs may occur at any
time during the infusion of Myozyme or generally up to 2 hours after, and are more likely with higher
infusion rates.
Patients with advanced Pompe disease may have compromised cardiac and respiratory function, which
may predispose them to a higher risk of severe complications from infusion associated reactions.
Therefore, these patients should be monitored more closely during administration of Myozyme.
Immunogenicity
In clinical studies, the majority of patients developed IgG antibodies to rhGAA typically within
3 months of treatment. Thus seroconversion is expected to occur in most patients treated with
Myozyme. A tendency was observed for infantile-onset patients treated with a higher dose (40 mg/kg)
3
to develop higher titers of antibodies. There does not appear to be a correlation between the onset of
IARs and the time of IgG antibody formation. A limited number of the IgG positive patients evaluated
tested positive for inhibitory effects on
in vitro
testing. Due to the rarity of the condition and the
limited experience to date, the effect of antibody formation on safety and efficacy is currently not
fully established. The probability of a poor outcome and of developing high and sustained antibody
titers appears higher among CRIM-negative patients (Cross Reactive Immunologic Material; patients
in whom no endogenous GAA protein was detected by Western blot analysis) than among CRIM-
positive patients (patients in whom endogenous GAA protein was detected by Western blot analysis).
However, high and sustained antibody titers also occur in some CRIM-positive patients. The cause of
a poor clinical outcome and of developing high and sustained antibody titers is thought to be multi-
factorial. IgG antibody titers should be regularly monitored.
Patients who experience hypersensitivity reactions may also be tested for IgE antibodies to
alglucosidase alfa and other mediators of anaphylaxis. Patients who develop IgE antibodies to
Myozyme appear to be at a higher risk for the occurrence of IARs when Myozyme is re-administered
(see section 4.8). Therefore, these patients should be monitored more closely during administration of
Myozyme. Some IgE positive patients were successfully rechallenged with Myozyme using a slower
infusion rate at lower initial doses and have continued to receive Myozyme under close clinical
supervision.
Transient nephrotic syndrome
Transient nephrotic syndrome which resolved following temporary interruption of ERT was observed
in one patient with infantile-onset Pompe disease who received very frequent dosing of rhGAA
(10 mg/kg 5 times weekly) over an extended period.
Immune complex-mediated reactions
Severe cutaneous reactions, possibly immune-mediated, have been reported with alglucosidase alfa,
including ulcerative and necrotizing skin lesions (see section 4.8). Patients should be monitored for
signs and symptoms of systemic immune complex-mediated reactions involving skin and other organs
while receiving alglucosidase alfa. If immune-mediated reactions occur, discontinuation of the
administration of alglucosidase alfa should be considered and appropriate medical treatment initiated.
The risks and benefits of re-administering alglucosidase alfa following an immune-mediated reaction
should be considered. Some patients have been successfully rechallenged and continued to receive
alglucosidase alfa under close clinical supervision.
No interactions studies have been performed. Because it is a recombinant human protein,
alglucosidase alfa is an unlikely candidate for cytochrome P450 mediated drug-drug interactions.
Pregnancy
There are no data from the use of alglucosidase alfa in pregnant women. Studies in animals have
shown reproductive toxicity (see section 5.3). The potential risk for humans is unknown.
Myozyme
should not be used during pregnancy unless clearly necessary.
Breast-feeding
Alglucosidase alfa may be excreted in breast milk. Because there are no data available on effects in
neonates exposed to alglucosidase alfa via breast milk, it is recommended to stop breast-feeding when
Myozyme is used.
Fertility
There are no clinical data on the effects of alglucosidase alfa on fertility. Preclinical data did not
reveal any significant adverse findings (see section 5.3).
4
No studies on the effects on the ability to drive and use machines have been performed. Because
dizziness has been reported as an infusion associated reaction, this may affect the ability to drive and
use machines on the day of the infusion.
Infantile-onset Pompe disease
In clinical trials, 39 infantile-onset patients were treated with Myozyme for more than three years
(168 weeks with a median of 121 weeks; see section 5.1). Adverse reactions reported in at least
2 patients are listed in Table 1 by System Organ Class. Adverse reactions were mostly mild to
moderate in intensity and almost all occurred during the infusion or during the 2 hours following the
infusion (infusion associated reactions, IARs). Serious infusion reactions including urticaria, rales,
tachycardia, decreased oxygen saturation, bronchospasm, tachypnea, periorbital edema and
hypertension have been reported.
Late-onset Pompe disease
In a placebo-controlled study lasting 78 weeks, 90 patients with late-onset Pompe disease, aged 10 to
70 years, were treated with Myozyme or placebo randomized in a 2:1 ratio (see section 5.1). Overall,
the numbers of patients experiencing adverse reactions and serious adverse reactions were comparable
between the two groups. The most common adverse reactions observed were IARs. Slightly more
patients in the Myozyme group than in the placebo group experienced IARs (28% versus 23%). The
majority of these reactions were non-serious, mild to moderate in intensity and resolved
spontaneously. Adverse reactions reported in at least 2 patients are listed in Table 1. Serious adverse
reactions reported in 4 patients treated with Myozyme were: angioedema, chest discomfort, throat
tightness, non-cardiac chest pain and supraventricular tachycardia. Reactions in 2 of these patients
were IgE-mediated hypersensitivity reactions.
Table 1: Adverse reactions (reported in at least 2 patients) and adverse reactions reported in post-
marketing setting, expanded access programs and non-controlled clinical trials, per System Organ
Class, presented by frequency categories: very common (≥1/10), common (≥1/100 to<1/10),
uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000) and not known
(cannot be estimated from the available data). Due to the small patient population, an adverse reaction
reported in 2 patients is classified as common. Within each frequency grouping, adverse reactions are
presented in order of decreasing seriousness.
System Organ
Class
Frequency
Adverse reaction
(Preferred Term Level)
Additional adverse
reactions
4
Infantile- and Late-
onset Pompe disease
Infantile-onset
Pompe disease
1
Late-onset
Pompe disease
2
Immune system
disorders
common
Hypersensitivity
Psychiatric
disorders
common
Agitation
not known
Agitation
Restlessness
Nervous system
disorders
common
Tremor
Dizziness
Paraesthesia
Headache
3
not known
Tremor
Headache
Eye disorders
not known
Conjunctivitis
Cardiac disorders very
Tachycardia
5
















common
common
Cyanosis
not known
Cardiac arrest
Bradycardia
Tachycardia
Cyanosis
Vascular
disorders
very
common
Flushing
common
Hypertension
Pallor
Flushing
not known
Hypertension
Hypotension
Pallor
Respiratory,
thoracic and
mediastinal
disorders
very
common
Tachypnoea
Cough
common
Throat tightness
not known
Respiratory arrest
Apnea
Respiratory distress
Bronchospasm
Wheezing
Pharyngeal oedema
Dyspnoea
Tachypnoea
Throat tightness
Cough
Gastrointestinal
disorders
very
common
Vomiting
common
Retching
Nausea
Diarrhoea
Vomiting
Nausea
3
not known
Abdominal pain
Retching
Skin and
subcutaneous
tissue disorders
very
common
Urticaria
Rash
common
Erythema
Rash maculopapular
Rash macular
Rash papular
Pruritus
Urticaria
Rash papular
Pruritus
Hyperhidrosis
not known
Periorbital edema
Livedo reticularis
Lacrimation increased
Rash
Erythema
Hyperhidrosis
Musculoskeletal
and connective
tissue disorders
common
Muscle spasms
Muscle twitching
Myalgia
not known
Arthralgia
General disorders
and
administration
site conditions
very
common
Pyrexia
common
Irritability
Chills
Pyrexia
Chest discomfort
6

























Peripheral oedema
Local swelling
Fatigue
3
Feeling hot
not known
Chest pain
Face edema
Feeling hot
Pyrexia
Chills
Chest discomfort
Irritability
Peripheral coldness
Infusion site pain
Infusion site reaction
Investigations
very
common
Oxygen saturation
decreased
common
Heart rate increased
Blood pressure
increased
Body temperature
increased
Blood pressure
increased
not known
Oxygen saturation
decreased
Heart rate increased
1
Reactions reported in 39 infantile-onset patients in 2 clinical trials
2
Reactions reported in 60 late-onset patients in a placebo-controlled clinical trial
3
Reactions reported more frequently in the placebo group than in the Myozyme group in late-onset patients
4
Additional adverse reactions from post-marketing, expanded access programs and non-controlled clinical trials.
A small number of patients (<1%) in clinical trials and in the commercial setting developed
anaphylactic shock and/or cardiac arrest during Myozyme infusion that required life-support
measures. Reactions generally occurred shortly after initiation of the infusion. Patients presented with
a constellation of signs and symptoms, primarily respiratory, cardiovascular, edematous and/or
cutaneous in nature (see section 4.4).
Patients with moderate to severe or recurrent IARs have been evaluated for Myozyme specific IgE
antibodies; some patients tested positive including some who experienced an anaphylactic reaction.
Severe cutaneous reactions, possibly immune-mediated, have been reported with alglucosidase alfa
including ulcerative and necrotizing skin lesions (see section 4.4).
There is no experience with overdose of alglucosidase alfa.
In clinical studies doses up to
40 mg/kg body weight were used.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other alimentary tract and metabolism products, enzymes.
ATC code: A16AB07.
7











Pompe disease
Pompe disease is a rare, progressive and fatal metabolic myopathy with an estimated global incidence
of 1 in 40,000 births. Other names for Pompe disease include glycogen storage disease type II (GSD-
II), acid maltase deficiency (AMD) and glycogenosis type II. Pompe disease belongs to the lysosomal
storage disorders as it is caused by a deficiency of a naturally-occurring lysosomal hydrolase, acid -
glucosidase (GAA) that degrades lysosomal glycogen to glucose. Deficiency of this enzyme leads to
glycogen accumulation in various tissues, particularly cardiac, respiratory and skeletal muscle, leading
to the development of hypertrophic cardiomyopathy and progressive muscle weakness, including
impairment of respiratory function.
The clinical presentation of Pompe disease can be described as a spectrum of disease which ranges
from a rapidly-progressing infantile-onset form (onset of symptoms of Pompe disease typically within
the first year of life and a very short expected life-span) to a less rapidly-progressing late-onset form.
The infantile-onset form of Pompe disease is characterised by massive deposition of glycogen in the
heart, and skeletal muscle always resulting in rapidly progressive cardiomyopathy, generalised muscle
weakness and hypotonia. Motor development is often completely arrested, or if motor milestones are
achieved, they are subsequently lost. Death typically occurs due to cardiac and/or respiratory failure
before the age of one year.
In a retrospective natural history study in patients with infantile-onset Pompe disease (n=168), the
median age at onset of symptoms was 2.0 months and the median age of death was 9.0 months.
Kaplan-Meier survival rates at 12, 24 and 36 months of age were 26%, 9% and 7%, respectively.
A non-typical, more slowly progressive form of infantile-onset Pompe disease has been described
which is characterised by a less severe cardiomyopathy and consequently a more prolonged survival.
The late-onset form of Pompe disease manifests during infancy, childhood, adolescence or even
adulthood and is much less rapidly progressive than the infantile-onset form. Usually, it is
characterised by the presence of sufficient residual GAA activity to preclude the development of
cardiomyopathy, however some cardiac involvement has been reported in up to approximately 4% of
patients with late-onset Pompe disease.
Patients with late-onset Pompe disease typically present with progressive myopathy, predominantly of
the proximal muscles in the pelvic and shoulder girdles, and varying degrees of respiratory
involvement, ultimately progressing to profound disability and/or the need for ventilatory support.
The time course of disease progression is extremely variable and not predictable, with some patients
experiencing a rapid deterioration in skeletal and respiratory muscle function leading to loss of
ambulation and respiratory failure, others progressing less rapidly, and yet others presenting with a
dissociation in the progression of skeletal and respiratory muscle involvement.
It is postulated that Myozyme will restore lysosomal GAA activity resulting in stabilisation or
restoration of cardiac and skeletal muscle function (including respiratory muscles). Due to the blood-
brain barrier effect and the enzyme‟s size, uptake of alglucosidase alfa in the central nervous system is
unlikely.
Infantile-onset Pompe disease; clinical trial in patients aged 6 months or less
The safety and efficacy of Myozyme was assessed in a pivotal, randomised, open-label, historically-
controlled clinical trial of 18 non-ventilated infantile-onset patients aged 6 months or less at the onset
of treatment. The untreated historical cohort was matched to the pivotal study population and was
derived from a retrospective natural history study (n=42) in patients with infantile-onset Pompe
disease. Patients were randomized to receive either 20 mg/kg or 40 mg/kg once every two weeks for a
period of 52 weeks. After a minimum of 52 weeks, 16 of these 18 patients were enrolled in an
extension study to receive continued treatment at the same dose for a total duration of up to three
years (150 weeks).
8
The primary endpoint was the proportion of patients who were alive and free of invasive ventilator
support.
However, the invasive ventilator-free survival was not recorded in the untreated historical
cohort and a comparison of this endpoint is not possible. After 52 weeks of treatment, all 18 patients
treated with Myozyme were alive and 15 of these 18 patients were alive and free of invasive
ventilatory support whereas 1 of 42 patients in the untreated historical cohort was alive at 18 months
of age. Two patients died and did not enter into the extension study. After 104 weeks of treatment, all
16 patients who enrolled in the extension study were alive and 10 of these 16 patients were free of
invasive ventilatory support. At the end of the study (with individual patient treatment durations
ranging from 60 to 150 weeks; mean follow-up period of 119 weeks) 14 of 16 patients were alive and
9 of 16 patients were alive and free of invasive ventilatory support. One additional patient died after
study end and another one after withdrawal from the study.
Comparison of survival curves from time of diagnosis versus the untreated historical cohort was made
using a Cox proportional hazards regression analysis. Patients treated with Myozyme demonstrated
prolonged survival as compared to survival in an untreated historical cohort (see Table 2).
Table 2: Results for endpoint survival using the Cox regression model
Treated
Patients
Historical
Reference
Comparator
Endpoint
Treatment
Effect Hazard
Ratio
95%
Confidence
Interval
p-value
<0.0001
Note: Results are from a Cox proportional hazards regression analysis which includes treatment
as a time-varying covariate, and also includes age of diagnosis and age at symptom onset.
Subjects were aged 6 months or less at the onset of treatment.
Subjects in the untreated historical cohort were born in 1993 or later.
N=42
Survival
0.05
(0.015, 0.147)
Echocardiographic indices of cardiomyopathy improved as measured by a decrease in left ventricular
mass (LVM). After 52 weeks of treatment, LVM decreased from baseline in all 14 patients with
available data and was within normal limits in 3 of 14 patients. After the first year (64 up to
130 weeks) of treatment LVM further decreased in 8 patients. At 104 weeks of treatment LVM
assessments were available for 8 patients, of which 5 decreased to within normal limits.
As measured by motor performance age-equivalent scores of the Alberta Infant Motor Scale (AIMS),
seven of the 18 patients made motor development gains during the study and were walking
independently by the last study assessment (with individual patient treatment durations ranging from
52 to 130 weeks; mean follow-up period of 94 weeks). An additional 4 patients made motor
development gains during the study and were sitting independently by the last study assessment (with
individual patient treatment durations ranging from 78 to 130 weeks; mean follow-up period of
110 weeks), although they did not have functional use of the legs. The remaining 7 patients made no
clinically significant motor gains or were unable to sustain the motor gains made and had very limited
motor movement by the last study assessment (with individual patient treatment durations ranging
from 52 to 142 weeks; mean follow-up period of 103 weeks).
After 52 weeks of treatment 14 of 18 patients (77.8%) had maintained or improved weight-for-age
percentiles (above the 3rd percentile), 14 of 15 patients (93.3%) were above the 3rd percentile for
length and 12 of 15 patients (80.0%) were above the 3rd percentile for head circumference. In the
second year of treatment, 15 out of 17 patients had further improved weight-for-age percentiles (with
individual patient treatment durations ranging from 78 to 142 weeks; mean follow-up period of
111 weeks), 10 out of 16 patients had further improved length-for-age percentiles (with individual
patient treatment durations ranging from 90 to 130 weeks; mean follow-up period of 113 weeks) and
11 out of 15 patients had further improved head circumference-for-age percentiles (with individual
patient treatment durations ranging from 90 to 130 weeks; mean follow-up period of 110
weeks). At
104 weeks of treatment, all 13 patients with available data had maintained or improved weight-for-age
percentiles (above the 3rd percentile), all 12 patients with available data were above the 3rd percentile
9
N=18










for length and all 12 patients with available data were above the 3rd percentile for head
circumference.
Analyses of efficacy did not reveal meaningful differences between the 2 dose groups with respect to
survival, invasive ventilator-free survival, any ventilator-free survival, decrease in LVM, gains in
growth parameters and acquisition of motor milestones. Based on these results the 20 mg/kg qow dose
is recommended.
Infantile-onset Pompe disease; clinical trial in patients aged 6 months to 3.5 years
A second open-label clinical trial also assessed the safety and efficacy of Myozyme in 21 patients
with predominantly a non-typical form of infantile-onset Pompe disease who ranged in age from
6 months to 3.5 years at initiation of treatment. Patients received 20 mg/kg Myozyme once every two
weeks for 52 weeks except for 8 patients who received 40 mg/kg after at least 26 weeks of treatment.
After 52 weeks all patients continued treatment for a total duration of more than 3 years (168 weeks
with a median of 121 weeks).
The primary endpoint of the pivotal trial was the proportion of patients who were alive. After
52 weeks of treatment, 16 of 21 patients (76.2%) treated with Myozyme were alive. After 104 weeks
of treatment, 14 of 21 patients (66.7%) were alive and 1 patient was alive but had discontinued from
the study. These proportions were maintained up to the end of the study (with individual patient
treatment durations ranging from 1 to 168 weeks; mean follow-up period of 109 weeks). In the
untreated historical cohort 5 of 47 patients (10.6%) for whom data were available, were alive at age
30 months (2.5 years).
Survival in the treated patients was compared to survival in a similar historical cohort of untreated
subjects using a Cox proportional hazards regression analysis (See Table 3).
Table 3: Results for endpoint survival using the Cox regression model
Treated
Patients
Historical
Reference
Comparator
Endpoint
Treatment
Effect Hazard
Ratio
95%
Confidence
Interval
p-value
0.0166
Note: Results are from a Cox proportional hazards regression analysis which includes treatment
as a time-varying covariate, and also includes age of diagnosis and age at symptom onset.
Subjects ranged in age from 6 months to 3.5 years at initiation of treatment.
Subjects in the untreated historical cohort were born in 1995 or later.
N=48
Survival
0.301
(0.112,0.804)
Additional efficacy data showed that of 16 patients who were free of invasive-ventilator support at
baseline, 7 remained so after 104 weeks of treatment. The 9 remaining patients either died (5 patients)
or became invasive-ventilator dependent (4 patients). All 5 patients who were receiving invasive
ventilation at baseline continued to require ventilation throughout the study (4 patients survived
beyond week 104 and one patient died).
After 52 weeks of treatment, LVM decreased from baseline in all 12 patients with available data and
was within normal limits in 6 of 12 patients. After the first year (58 up to 168 weeks) of treatment
LVM further decreased in 9 out of 12 patients with available data. At 104 weeks of treatment LVM
assessments were available for 10 patients, of which 9 decreased to within normal limits.
After 52 weeks of treatment, 3 out of 8 patients with available data made gains in motor function over
baseline as measured by raw scores and age-equivalent scores from baseline in the AIMS. Six of the
11 patients with available data continued to make motor development gains beyond Week 52 (with
individual patient treatment durations ranging from 58 to 168 weeks; mean follow-up period of
121 weeks), including 3 patients ambulatory and 3 patients with only functional sitting skills by the
last study visit. The remaining 5 patients showed no significant change in motor development beyond
Week 52 (with individual patient treatment durations ranging from 104 to 168 weeks; mean follow-up
10
N=21










period of 140 weeks), including 4 patients with no significant motor skills in any of the positions
evaluated and 1 patient with only functional sitting skills by the last study visit.
The vast majority of patients with infantile-onset Pompe disease treated with Myozyme demonstrate
improvement in cardiac function as well as stabilisation or improvements in growth parameters.
However, motor and respiratory responses to treatment have been more variable.
Patients with infantile-onset Pompe disease who demonstrated motor gains, had greater preservation
of motor function and lower glycogen content in the quadriceps muscle at baseline. It is noteworthy
that a higher proportion of patients with better motor outcomes show stability or improvement in
growth parameters (weight), while the large majority of patients, regardless of their motor outcomes
or baseline features, show reversal of cardiomyopathy as measured by changes in LVM Z-score.
The totality of the data suggests that early diagnosis and treatment at an early stage of disease may be
critical to achieve the best outcomes in these infantile onset patients.
Late-onset Pompe disease
The safety and efficacy of Myozyme was assessed in a randomized, double-blind, placebo-controlled
study in 90 patients with late-onset Pompe disease who ranged in age from 10 to 70 years at initiation
of treatment and were all naive to enzyme replacement therapy. Patients were randomized in a 2:1
ratio and received 20 mg/kg Myozyme (n=60) or placebo (n=30) once every two weeks for 78 weeks
(18 months).
The co-primary efficacy outcome assessments were distance walked (meters) in 6 minutes (6-Minute
Walk Test, 6MWT) and FVC (Forced Vital Capacity) % predicted in the sitting position. After
78 weeks, patients treated with Myozyme showed improvement in distance walked as measured by
6MWT and stabilization of pulmonary function as measured by FVC % predicted as compared to
placebo-treated patients. The distance walked in 6 minutes increased by a median of 15.0 meters for
Myozyme-treated patients and decreased by a median of 7.5 meters for placebo-treated patients,
indicating a statistically significant Myozyme treatment effect compared to placebo (p=0.0283). The
% predicted FVC changed by a median of 0.0 for Myozyme-treated patients and decreased by a
median of 3% for placebo-treated patients, indicating a statistically significant treatment effect
(p=0.0026). The results are shown in Table 4.
11
Table 4: Change from baseline: efficacy outcomes in the placebo-controlled study
Myozyme
(N = 60)
Placebo
(N = 30)
6-Minute Walk Test Distance (meters)
Pre-treatment Baseline
Mean ± s.d.
Median
332.20 ± 126.69
360.0
317.93 ± 132.29
339.0
Week 78/Last Observation
Mean ± s.d.
Median
357.85 ± 141.32
367.5
313.07 ± 144.69
307.0
Change from Baseline to
Week 78/Last Observation*
Mean ± s.d
Median
26.08 ± 64.41
15.0
-4.87 ± 45.24
-7.5
Wilcoxon-Mann-Whitney Test
p-value
0.0283
Forced Vital Capacity (Percent of predicted normal)
Pre-treatment Baseline
Mean ± s.d.
Median
55.43 ± 14.44
53.5
53.00 ± 15.66
49.0
Week 78/Last Observation
Mean ± s.d.
Median
56.67 ± 16.17
55.5
50.70 ± 14.88
49.0
Change from Baseline to Week
78/Last Observation*
Mean ± s.d
Median
1.25 ± 5.55
0.0
-2.3 ± 4.33
-3.0
Wilcoxon-Mann-Whitney Test
p-value
0.0026
*One patient who did not have data post baseline was excluded from the analyses.
An open-label clinical trial assessed the safety and efficacy of Myozyme in 5 patients with late-onset
Pompe disease who ranged in age from 5 to 15
years at initiation of treatment. Patients received
20 mg/kg Myozyme once every two weeks for 26 weeks.
All patients were freely ambulatory and all
but one patient did not require any form of ventilator support (1 patient required nocturnal non-
invasive ventilation). Of the 3 patients with significant pulmonary involvement at screening/baseline
(percentage predicted forced vital capacity in the sitting position ranging from 58-67%), two
demonstrated clinically meaningful improvements in FVC (+11.5% and +16.0%) in the sitting
position by Week 26. Evaluation of motor function gave disparate results.
Ten patients with advanced late-onset Pompe disease (i.e. wheelchair-bound for 10/10 and ventilator-
dependent for 9/10) aged 9-54 years were treated in expanded access programs with alglucosidase alfa
20-40 mg/kg once every two weeks for various periods of time between 6 months and 2.5 years. The
pulmonary benefits observed in patients included a clinically meaningful improvement in FVC of
35% in one patient, and significant reductions in the number of hours of ventilator support needed in
2 patients. Benefits of treatment on motor function including the regaining of lost motor skills were
observed in some patients. Only one patient became wheelchair-free. In this group of patients a
variable response has also been seen with respect to motor function.
Pompe Registry
Medical or healthcare professionals are encouraged to register patients who are diagnosed with Pompe
disease at www.PompeRegistry.com. Patient data will be anonymously collected in this Registry. The
objectives of the “Pompe Registry” are to enhance the understanding of Pompe disease and to monitor
patients and their response to enzyme replacement therapy over time, with the ultimate goal of
improving clinical outcomes for these patients.
5.2 Pharmacokinetic properties
Infantile-onset Pompe disease
In a pivotal trial including 18 patients, the pharmacokinetics of alglucosidase alfa were evaluated in
15 patients with infantile-onset Pompe disease (all less than 6 months of age at treatment-onset) who
received doses of 20 mg/kg or 40 mg/kg alglucosidase alfa as an approximate 4 to 6.5-hour infusion,
12

















respectively. Pharmacokinetics were dose proportional and did not change over time. After the first
and sixth infusion of Myozyme, mean maximum plasma concentrations (C
max
) ranged from 178.2 to
263.7 g/ml for the 20 mg/kg and 40 mg/kg dose groups respectively. The mean area under the plasma
concentration-time curve (AUC ) ranged from 977.5 to 1,872.5 g h/ml for the 20 mg/kg and
40 mg/kg dose groups. Mean plasma clearance (CL) was 21.4 ml/h/kg and mean volume of
distribution at steady state (Vss) was 66.2 ml/kg for both dose groups with small between-subject
variability of 15% and 11%, respectively. Mean plasma elimination half-life (t
1/2
) was 2.75 hours for
the two dose groups.
The pharmacokinetics of alglucosidase alfa were also evaluated in a separate trial in 21 patients with
infantile-onset Pompe disease (all aged between 6 months and 3.5 years at treatment-onset) who
received doses of 20 mg/kg of alglucosidase alfa. In 12 patients with available data the AUC and
C
max
were approximately equivalent to those observed for the 20 mg/kg dose group in the pivotal trial.
The t½ of approximately 2-3 hours was also similar in this group of patients.
Late-onset Pompe disease
The pharmacokinetics of alglucosidase alfa were evaluated in a trial in 5 patients with late-onset
Pompe disease aged 6-15 years who received 20 mg/kg alglucosidase alfa once every two weeks.
There was no difference in the pharmacokinetic profile of alglucosidase alfa in these juvenile late-
onset patients compared to infantile-onset patients.
The pharmacokinetics of alglucosidase alfa were studied in a population analysis of 32 late-onset
Pompe disease patients from the randomized, double-blind, placebo-controlled study ranging in age
from 21 to 70 years who received Myozyme 20 mg/kg once every two weeks. AUC and C
max
were
similar at week 0, 12 and 52 visits indicating alglucosidase alfa pharmacokinetics were not time-
dependent (Table 5).
Table 5: Alglucosidase alfa pharmacokinetics after a single dose and after 12 and 52 weeks of
therapy
Parameter
Week 0
Week 12
Week 52
C
max
( g/ml)
385 106
349 79
370 88
AUC ( g h/ml)
2672 1140
2387 555
2700 1000
CL (ml/h/kg)
8.1 1.8
8.9 2.3
8.2 2.4
Vss (ml/kg)
904 1158
919 1154
896 1154
Effective half-life (h)
2.4 0.4
2.4 0.3
2.5 0.4
There was no evidence that IgG antibodies to alglucosidase alfa affected pharmacokinetics. Higher
mean clearance, lower mean AUC, and lower mean C
max
were observed in 5 patients who tested
positive for inhibition of cellular uptake of enzyme. However, there was no apparent association
between inhibition of uptake and the co-primary efficacy endpoints (see section 4.4).
5.3 Preclinical safety data
Preclinical data reveal no special hazards for humans based on studies of safety pharmacology, single
and repeat dose toxicity. No significant adverse findings on embryofoetal development were observed
in a mouse and a rabbit embryofoetal study and no significant adverse findings were observed in a
mouse fertility and early embryonic development study. In the rabbit embryofoetal development
study, following administration of Myozyme (10-40 mg/kg/day) with coadministration of
diphenhydramine, a treatment-related increase in the incidence of abortions and early delivery was
observed. This effect was partly attributable to maternal toxicity, as a significant decrease in feed
consumption and body weight gain was observed.
13


































6.
6.1 List of excipients
Mannitol
Sodium dihydrogen phosphate monohydrate
Disodium phosphate heptahydrate
Polysorbate 80
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3 Shelf life
2 years
After dilution, an immediate use is recommended. However, chemical and physical in-use stability has
been demonstrated for 24 hours at 2 to 8 C when stored under protection from light.
6.4 Special precautions for storage
Store in a refrigerator (2 C - 8 C).
For storage conditions of the diluted medicinal product, see section 6.3.
6.5
Nature and contents of container
50 mg of powder in a vial (Type 1 glass) with a stopper (siliconised butyl) and a seal (aluminium)
with a flip-off cap (plastic). Pack sizes of 1, 10 or 25 vials.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
Myozyme has to be reconstituted with water for injections, then diluted with sodium chloride 9 mg/ml
(0.9%) solution for injection and then administered by intravenous infusion. Reconstitution and
dilution should be performed in accordance with good practice rules, particularly for the respect of
asepsis.
Due to the proteinaceous nature of the product, particle formation may occur in the reconstituted
solution and final infusion bags. Therefore, a 0.2 micron low protein binding in-line filter should be
used for administration. It was demonstrated that the use of a 0.2 micron in-line filter removes visible
particles and does not result in an apparent loss of protein or activity.
Determine the number of vials to be reconstituted based on the individual patient’s dose regimen
(mg/kg) and remove the required vials from the refrigerator in order to allow them to reach room
temperature (approximately 30 minutes). Each vial of Myozyme is for single use only.
Use aseptic technique
Reconstitution
Reconstitute each 50 mg vial of Myozyme with 10.3 ml water for injections. Add the water for
injections by slow drop-wise addition down the side of the vial and not directly onto the lyophilised
14



cake. Tilt and roll each vial gently. Do not invert, swirl or shake the vial. The reconstituted volume is
10.5 ml containing 5 mg/ml, and appears as a clear, colourless to pale yellow solution which may
contain particles in the form of thin white strands or translucent fibres. Perform an immediate
inspection of the reconstituted vials for particulate matter and discoloration. If upon immediate
inspection foreign particles other than those described above are observed, or if the solution is
discoloured, do not use. The pH of the reconstituted solution is approximately 6.2.
After reconstitution, it is recommended to
promptly dilute
the vials (see below).
Dilution
When reconstituted as above, the reconstituted solution in the vial contains 5 mg alglucosidase alfa
per ml. The reconstituted volume allows accurate withdrawal of 10.0 ml (equal to 50 mg) from each
vial. This should then be further diluted as follows: Slowly withdraw the reconstituted solution from
each vial until the volume for the patient‟s dose is obtained. The recommended final concentration of
alglucosidase in the infusion bags ranges from 0.5 mg/ml to 4 mg/ml. Remove airspace within the
infusion bag. Also remove an equal volume of sodium chloride 9 mg/ml (0.9%) solution for injection,
that will be replaced with reconstituted Myozyme. Slowly inject the reconstituted Myozyme directly
into the sodium chloride 9 mg/ml (0.9%) solution for injection. Gently invert or massage the infusion
bag to mix the diluted solution. Do not shake or excessively agitate the infusion bag.
The final infusion solution should be administered as close to preparation time as possible.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Genzyme Europe B.V., Gooimeer 10, NL-1411 DD Naarden, The Netherlands
8.
EU/1/06/333/001-003
9.
Date of first authorisation:
29 March 2006
Date of latest renewal:
29 March 2011
Detailed information on this medicinal product is available on the website of the European Medicines
Agency
http://www.ema.europa.eu.
15
ANNEX II
A.
MANUFACTURERS OF THE BIOLOGICAL ACTIVE
SUBSTANCE AND MANUFACTURING AUTHORISATION
HOLDER RESPONSIBLE FOR BATCH RELEASE
B.
CONDITIONS OF THE MARKETING AUTHORISATION
16
A. MANUFACTURERS OF THE BIOLOGICAL ACTIVE SUBSTANCE AND
MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturers of the biological active substance
Genzyme Corp. 45, 51, 76, 74 and 80 New York Avenue, Framingham, MA 01701, USA
Genzyme Flanders bvba, Cipalstraat 8, 2440 Geel, Belgium
Name and address of the manufacturer responsible for batch release
Genzyme Ltd., 37 Hollands Road, Haverhill, Suffolk CB9 8PU, United Kingdom
Genzyme Ireland Ltd., IDA Industrial Park, Old Kilmeaden Road, Waterford, Ireland
The printed package leaflet of the medicinal product must state the name and address of the
manufacturer responsible for the release of the concerned batch.
B. CONDITIONS OF THE MARKETING AUTHORISATION
Medicinal product subject to restricted medical prescription (See Annex I: Summary of Product
Characteristics, section 4.2).
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND
EFFECTIVE USE OF THE MEDICINAL PRODUCT
Not appliable
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, presented in Module 1.8.1. of the
Marketing Authorisation, is in place and functioning before and whilst the product is on the market.
Risk Management Plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in
the Pharmacovigilance Plan, as agreed in version 4 of the Risk Management Plan (RMP) presented in
Module 1.8.2. of the Marketing Authorisation Application and any subsequent updates of the RMP
agreed by the CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, the
updated RMP should be submitted at the same time as the next Periodic Safety Update Report
(PSUR).
In addition, an updated RMP should be submitted
In addition, an updated RMP should be submitted
When new information is received that may impact on the current Safety Specification,
Pharmacovigilance Plan or risk minimisation activities
Within 60 days of an important (pharmacovigilance or risk minimisation) milestone being
reached
At the request of the European Medicines Agency.
17






ANNEX III
LABELLING AND PACKAGE LEAFLET
18
A. LABELLING
19
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
CARTON
1.
Myozyme 50 mg powder for concentrate for solution for infusion
Alglucosidase alfa
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each vial contains 50 mg of alglucosidase alfa.
After reconstitution, the solution contains 5 mg of alglucosidase alfa/ml and after dilution, the
concentration varies from 0.5 mg to 4 mg/ml.
3.
LIST OF EXCIPIENTS
Excipients:
Mannitol
Sodium dihydrogen phosphate monohydrate
Disodium phosphate heptahydrate
Polysorbate 80
See leaflet for further information.
4.
1 vial of powder for concentrate for solution for infusion.
10 vials of powder for concentrate for solution for infusion.
25 vials of powder for concentrate for solution for infusion.
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
For single use only
Read the package leaflet before use.
Intravenous use
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
20



























EXP
After dilution, an immediate use is recommended. However, chemical and physical in-use stability has
been demonstrated for 24 hours at 2 to 8 C when stored under protection from light.
9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator (at 2°C-8°C).
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Any unused product should be discarded.
Genzyme Europe B.V.
Gooimeer 10
NL-1411 DD Naarden
The Netherlands
EU/1/06/333/001
EU/1/06/333/002
EU/1/06/333/003
13. BATCH NUMBER
Batch
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Justification for not including Braille accepted
21























MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
VIAL LABEL
1.
Myozyme 50 mg powder for concentrate for solution for infusion
Alglucosidase alfa
Intravenous use after reconstitution and dilution
2.
METHOD OF ADMINISTRATION
Read the package leaflet before use.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Batch
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
50 mg
6.
OTHER
Store in a refrigerator (at 2°C-8°C).
Genzyme Europe B.V.-NL
22

















B. PACKAGE LEAFLET
23
PACKAGE LEAFLET: INFORMATION FOR THE USER
Myozyme 50 mg powder for concentrate for solution for infusion
Alglucosidase alfa
Read all of this leaflet carefully before you start using this medicine.
-
Keep this leaflet. You may need to read it again.
-
If you have any further questions, ask your doctor or pharmacist.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even if
their symptoms are the same as yours.
-
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor or pharmacist.
In this leaflet
:
1. What Myozyme is and what it is used for
2. Before you are given Myozyme
3. How Myozyme is given
4. Possible side effects
5.
How to store Myozyme
6.
1.
WHAT MYOZYME IS AND WHAT IT IS USED FOR
Myozyme is used to treat adults, children and adolescents of all ages who have a confirmed diagnosis
of Pompe disease.
People with Pompe disease have low levels of an enzyme called -glucosidase. This enzyme helps the
body control levels of glycogen (a type of carbohydrate). Glycogen provides the body with energy, but
in Pompe disease the levels can get too high.
Myozyme is an artificial enzyme called alglucosidase alfa – this can replace the natural enzyme which
is lacking in Pompe disease.
In patients with late-onset Pompe disease (typically a more slowly progressive form of Pompe disease
with onset of symptoms after infancy) the evidence of efficacy is limited.
2.
BEFORE YOU ARE GIVEN MYOZYME
You should not be given Myozyme
If you have experienced life-threatening allergic (hypersensitive) reactions to alglucosidase alfa or any
of the other ingredients of Myozyme and re-administration of the medicine was not successful.
Symptoms of life-threatening allergic reactions include low blood pressure, very fast heart rate,
difficulty breathing, vomiting, facial swelling, hives or rash.
Take special care with Myozyme
If you are treated with Myozyme, you may experience a so-called infusion-associated reaction while
you are being given the medicine or during the next 2 hours. Such a reaction comprises different
symptoms like feeling hot, chills, headache, dizziness, itchy skin and nausea (see section 4 for an
overview of all infusion-associated reactions). An infusion-associated reaction can sometimes be very
severe. If you experience a reaction like this, you should
tell your doctor immediately
. You may
need to be given additional medicines to prevent an allergic reaction (e.g. antihistamines and/or
corticosteroids) or to reduce fever (antipyretics).
24
Further information
If you experience severe ulcerative lesions of your skin, please inform your doctor. Your doctor
should consider discontinuation of the administration of Myozyme and initiate appropriate medical
treatment. Your doctor should consider the risks and benefits of re-administering Myozyme.
Using other medicines
Please tell your doctor if you are taking or have recently taken any other medicines, including
medicines obtained without a prescription.
Pregnancy and breast-feeding
There is no experience of the use of Myozyme in pregnant women. You should not be given
Myozyme during pregnancy unless clearly necessary. You are recommended to stop breast-feeding
when you are given Myozyme. Ask your doctor or pharmacist for advice before taking any medicine.
Driving and using machines
Take care when driving or using any tools or machines shortly after infusion of Myozyme, since you
may experience dizziness.
Important information about some of the ingredients of Myozyme
This medicine contains less than 1 mmol sodium (23 mg) per vial, i.e. essentially „sodium free‟.
3.
HOW MYOZYME IS GIVEN
Myozyme will be given to you under the supervision of a doctor who is experienced in the treatment
of Pompe disease.
The dose you receive is based on your body weight. The recommended dosage of Myozyme is 20 mg
per kg of body weight. It will be given to you once every 2 weeks.
Use in children
The recommended dosage of Myozyme in children is the same as in adults.
Instructions for proper use
Myozyme is given through a drip into a vein (by intravenous infusion). It is supplied as a powder
which will be mixed with sterile water before it is given.
If you are givenmore Myozyme than needed
There is no experience with overdose of Myozyme.
If you miss an infusion of Myozyme
If you have missed an infusion, please contact your doctor.
If you have any further questions on the use of this product, ask your doctor or pharmacist.
4.
POSSIBLE SIDE EFFECTS
Like all medicines, Myozyme can cause side effects, although not everybody gets them.
Side effects were mainly seen while patients were being given the medicine or shortly after (“infusion
related effects”). Some of these infusion related side effects became serious. Life threatening
reactions, including very severe generalised allergic reactions and anaphylactic shock, have been
reported in some patients. Symptoms of such reactions include low blood pressure, very fast heart
rate, difficulty breathing, vomiting, facial swelling, hives or rash. Should you experience any reaction
like this, please
tell your doctor immediately
. You may need to be given additional medicines to
25
prevent an allergic reaction (e.g. antihistamines and/or corticosteroids) or to reduce fever
(antipyretics).
The frequency of possible side effects listed below is defined using the following convention:
Very common:
affects more than 1 user in 10
Common:
affects 1 to 10 users in 100
Uncommon:
affects 1 to 10 users in 1,000
Rare:
affects 1 to 10 users in 10,000
Very rare:
affects less than 1 user in 10,000
Not known:
frequency cannot be estimated from the available data.
Very common:
Hives
Rash
Increased heart rate
(Facial) flushing
Fever or increased body temperature
Cough
Increased breathing rate
Vomiting
Low level of oxygen in the blood
Common:
Paleness
Increased or high blood pressure
Bluish discolouration of the skin
Chills
Agitation
Tremor
Headache
Tingling
Pain or local reaction at the site of the drip
Dizziness
Irritability
Itchy skin
Retching
Swelling of the face, swelling of the throat or severe combined swelling of the face, throat and
tongue due to a severe allergic reaction
Swelling of the arms and legs
Nausea
Chest discomfort
Throat tightness
Diarrhoea
Tiredness
Muscle pain
Muscle spasms
Severe ulcerative lesions of the skin
Redness of the skin
Not known:
Swelling around the eyes
Abnormal breathing sounds
26













































Difficulty in breathing (including shortness of breath)
Cold extremities (e.g. hands, feet)
Low blood pressure
Sudden constriction of bronchi restricting air going in and out the lungs
Feeling hot
Increased sweating
Eyes tearing
Mottled skin
Restlessness
Wheezing
Decreased heart rate
Heart stopping
Chest pain (not in the heart)
Inflammation of membrane that covers eyeball and eyelid
Abdominal pain
Joint pain
Temporary suspension or sudden cessation of breathing
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet, please
tell your doctor or pharmacist.
5.
HOW TO STORE MYOZYME
Keep out of the reach and sight of children
You should not be given Myozyme after the expiry date which is stated on the labelling after ‘EXP’.
The expiry date refers to the last day of that month.
Store in a refrigerator (2°C – 8°C).
After dilution, an immediate use is recommended. However, chemical and physical in-use stability has
been demonstrated for 24 hours at 2 to 8 C when stored under protection from light.
Medicines should not be disposed of via wastewater or household waste. Ask your pharmacist how to
dispose of medicines no longer required. These measures will help to protect the environment.
6.
FURTHER INFORMATION
What Myozyme contains
-
The active substance is alglucosidase alfa. One vial contains 50 mg of alglucosidase alfa. After
reconstitution, the solution contains 5 mg of alglucosidase alfa per ml and after dilution, the
concentration varies from 0.5 mg to 4 mg/ml.
-
The other ingredients are
-
mannitol,
-
sodium dihydrogen phosphate monohydrate
-
disodium phosphate heptahydrate
-
polysorbate 80
What Myozyme looks like and contents of the pack
Myozyme is a powder for concentrate for solution for infusion in a vial (50 mg/vial). Each pack
contains 1, 10 or 25 vials. Not all pack sizes may be marketed.
27


















The powder is white to off-white. After reconstitution it is a clear, colourless to pale yellow solution,
which may contain particles. The reconstituted solution must be further diluted.
Marketing Authorisation Holder and Manufacturer
Marketing Authorisation Holder
Genzyme Europe B.V., Gooimeer 10, 1411 DD, Naarden, The Netherlands
Manufacturer
Genzyme Ltd., 37 Hollands Road, Haverhill, Suffolk CB9 8PU, United Kingdom
Genzyme Ireland Ltd., IDA Industrial Park, Old Kilmeaden Road, Waterford, Ireland
28
For any information about this medicine, please contact the local representative of the Marketing
Authorisation Holder:
België/Belgique/Belgien/
Luxemburg/Luxembourg
Genzyme Belgium N.V.,
Tel/ Tél: +32 2 714 17 11
Italia/Malta
Genzyme Srl (Italia/Italja),
Tel: +39 059 349811
България
Търговско представителство на
Genzyme CEE GmbH,
Тел.: +359 2 971 1001
Magyarország
Genzyme Europe B.V. Képviselet,
Tel: +36 1 310 7440
Česká Republika/Slovenská Republika/
Slovenija
Genzyme Czech s.r.o.,
Tel: +420 221 722 511
Nederland
Genzyme Europe B.V.,
Tel: +31 35 699 1200
Danmark/Norge/Sverige/Suomi/Finland/
Ísland
Genzyme A/S, (Danmark/Tanska/Danmörk),
Tlf/Puh./Sími: +45 32 71 2600
Österreich
Genzyme Austria GmbH,
Tel: +43 1 774 65 38
Deutschland
Genzyme GmbH,
Tel: +49 610236740
Polska/Eesti/Latvija/Lietuva
Genzyme Polska Sp. z o.o.
(Poola/Polija/Lenkija),
Tel: +48 22 24 60 900
Ελλά/Κύπρς
Genzyme Hellas Ltd. (Ελλά),
Τηλ: +30 210 99 49 270
Portugal
Genzyme Portugal S.A.,
Tel: +351 21 422 0100
España
Genzyme, S.L.U.,
Tel: +34 91 6591670
România
Genzyme CEE GmbH- Reprezentanţa pentru
România,
Tel: +40.21.243.42.28
France
Genzyme S.A.S.,
Tél: +33 (0) 825 825 863
United Kingdom/Ireland
Genzyme Therapeutics Ltd (United
Kingdom),
Tel: +44 1865 405200
This leaflet was last approved in
Detailed information on this medicine is available on the European Medicines Agency web site:
http://www.ema.europa.eu. There are also links to other websites about rare diseases and treatments.
<-----------------------------------------------------------------------------------------------------------------------------
The following information is intended for medical or healthcare professionals only:
29
Instructions for use – reconstitution, dilution and administration
Myozyme has to be reconstituted with water for injections, then diluted with sodium chloride 9 mg/ml
(0.9%) solution for injection and then administered by intravenous infusion. Reconstitution and
dilution should be performed in accordance with good practice rules, particularly for the respect of
asepsis.
Due to the proteinaceous nature of the product, particle formation may occur in the reconstituted
solution and final infusion bags. Therefore, a 0.2 micron low protein binding in-line filter should be
used for administration. It was demonstrated that the use of a 0.2 micron in-line filter removes visible
particles and does not result in an apparent loss of protein or activity.
Determine the number of vials to be reconstituted based on the individual patient’s dose regimen
(mg/kg) and remove the required vials from the refrigerator in order to allow them to reach room
temperature (approximately 30 minutes). Each vial of Myozyme is for single use only.
Use aseptic technique
Reconstitution
Reconstitute each 50 mg vial of Myozyme with 10.3 ml water for injections using a syringe with a
needle diameter not larger than 20 gauge. Add the water for injections by slow drop-wise addition
down the side of the vial and not directly onto the lyophilised cake. Tilt and roll each vial gently. Do
not invert, swirl or shake the vial. The reconstituted volume is 10.5 ml containing 5 mg enzyme/ml,
and appears as a clear, colourless to pale yellow solution which may contain particles in the form of
thin white strands or translucent fibres. Perform an immediate inspection of the reconstituted vials for
particulate matter and discoloration. If upon immediate inspection foreign particles other than those
described above are observed, or if the solution is discoloured, do not use. The pH of the reconstituted
solution is approximately 6.2.
After reconstitution it is recommended to
promptly dilute
the vials (see below).
Dilution
When reconstituted as above, the reconstituted solution in the vial contains 5 mg alglucosidase alfa
per ml. The reconstituted volume allows accurate withdrawal of 10.0 ml (equal to 50 mg) from each
vial. This should then be further diluted as follows: Slowly withdraw the reconstituted solution from
each vial until the volume for the patient’s dose is obtained using a syringe with a needle diameter not
larger than 20 gauge. The recommended final concentration of alglucosidase in the infusion bags
ranges from 0.5 mg/ml to 4 mg/ml. Remove airspace within the infusion bag. Also remove an equal
volume of sodium chloride 9 mg/ml (0.9%) solution for injection, that will be replaced with
reconstituted Myozyme. Slowly inject the reconstituted Myozyme directly into the sodium chloride
9 mg/ml (0.9%) solution for injection. Gently invert or massage the infusion bag to mix the diluted
solution. Do not shake or excessively agitate the infusion bag.
The final infusion solution should be administered as close to preparation time as possible.
Any unused product or waste material should be disposed of in accordance with local requirements.
Administration
It is recommended to start the administration of the diluted solution within three hours. The total time
between reconstitution and completion of the infusion should not exceed 24 hours.
The recommended dose regimen of Myozyme is 20 mg/kg of body weight administered once every 2
weeks as an intravenous infusion.
30



Infusions should be administered incrementally. It is recommended that the infusion begin at an initial
rate of 1 mg/kg/h and be gradually increased by 2 mg/kg/h every 30 minutes if there are no signs of
IARs (Infusion Associated Reactions) until a maximum rate of 7 mg/kg/h is reached.
31
Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/myozyme.html
Copyright © 1995-2021 ITA all rights reserved.