










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
NeoRecormon Multidose 50,000 IU Lyophilisate and solvent for solution for injection (5000 IU/ml)
2.
One vial contains 50,000 international units (IU) corresponding to 415 micrograms epoetin beta*
(recombinant human erythropoietin).
One ampoule contains 10 ml solvent (water for injections with benzyl alcohol and benzalkonium
chloride as preservatives).
One ml of reconstituted solution contains 5000 IU epoetin beta.
* produced in Chinese Hamster Ovary cells (CHO) by recombinant DNA technology
Excipients:
Phenylalanine (up to 5.0 mg/vial)
Sodium (less than 1 mmol per dose)
Benzyl alcohol (up to 40 mg per multidose solvent ampoule)
For a full list of excipients, see section 6.1.
3.
Lyophilisate and solvent for solution for injection.
White lyophilisate and clear, colourless solvent.
4.
-
Treatment of symptomatic anaemia associated with chronic renal failure (CRF) in adult and
paediatric patients.
-
Treatment of symptomatic anaemia in adult patients with non-myeloid malignancies receiving
chemotherapy.
-
Increasing the yield of autologous blood from patients in a pre-donation programme.
Its use in this indication must be balanced against the reported increased risk of thromboembolic
events. Treatment should only be given to patients with moderate anaemia (Hb 10 - 13 g/dl
[6.21 - 8.07 mmol/l], no iron deficiency) if blood conserving procedures are not available or
insufficient when the scheduled major elective surgery requires a large volume of blood (4 or
more units of blood for females or 5 or more units for males).
Therapy with NeoRecormon should be initiated by physicians experienced in the above mentioned
indications. As anaphylactoid reactions were observed in isolated cases, it is recommended that the
first dose be administered under medical supervision.
This multidose preparation can be used for several patients. To avoid the risk of cross-infection always
follow aseptic techniques and use disposable sterile syringes and needles for each administration.
Please check that only one vial of NeoRecormon Multidose is in use (i.e. reconstituted) at any one
time.
The reconstituted product is a colourless, clear to slightly opalescent solution.
For instructions on reconstitution of the product before administration, see section 6.6.
2
Treatment of symptomatic anaemia in adult and paediatric chronic renal failure patients:
Anaemia symptoms and sequelae may vary with age, gender, and overall burden of disease; a
physician’s evaluation of the individual patient’s clinical course and condition is necessary.
NeoRecormon should be administered either subcutaneously or intravenously in order to increase
haemoglobin to not greater than 12 g/dl (7.5 mmol/l). Subcutaneous use is preferable in patients who
are not receiving haemodialysis to avoid puncture of peripheral veins. In case of intravenous
administration, the solution should be injected over approx. 2 minutes, e.g. in haemodialysis patients
via the arterio-venous fistula at the end of dialysis.
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.2 mmol/l) to 12 g/dl (7.5 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.5 mmol/l) should be avoided; guidance for appropriate dose adjustment for when haemoglobin
values exceeding 12 g/dl (7.5 mmol/l) are observed are described below.
A rise in haemoglobin of greater than 2 g/dl (1.25 mmol/l) over a four week period should be avoided.
If it occurs, appropriate dose adjustment should be made as provided. If the rate of rise in haemoglobin
is greater than 2 g/dl (1.25 mmol/l) in one month or if the haemoglobin level is increasing and
approaching 12 g/dl (7.45 mmol/l), the dose is to be reduced by approximately 25 %. If the
haemoglobin level continues to increase, therapy should be interrupted until the hemoglobin level
begins to decrease, at which point therapy should be restarted at a dose approximately 25 % below the
previously administered dose.
Patients should be monitored closely to ensure that the lowest approved dose of NeoRecormon is used
to provide adequate control of the symptoms of anaemia.
In the presence of hypertension or existing cardiovascular, cerebrovascular or peripheral vascular
diseases, the weekly increase in Hb and the target Hb should be determined individually taking into
account the clinical picture.
Treatment with NeoRecormon is divided into two stages.
1.
Correction phase
- Subcutaneous administration:
- The initial dosage is 3 x 20 IU/kg body weight per week. The dosage may be increased every 4
weeks by 3 x 20 IU/kg and week if the increase of Hb is not adequate (< 0.25 g/dl per week).
- The weekly dose can also be divided into daily doses.
- Intravenous administration:
The initial dosage is 3 x 40 IU/kg per week. The dosage may be raised after 4 weeks to 80 IU/kg
three times per week - and by further increments of 20 IU/kg if needed, three times per week, at
monthly intervals.
For both routes of administration, the maximum dose should not exceed 720 IU/kg per week.
2.
Maintenance phase
To maintain an Hb of between 10 and 12 g/dl, the dosage is initially reduced to half of the previously
administered amount. Subsequently, the dose is adjusted at intervals of one or two weeks individually
for the patient (maintenance dose).
In the case of subcutaneous administration, the weekly dose can be given as one injection per week or
in divided doses three or seven times per week. Patients who are stable on a once weekly dosing
regimen may be switched to once every two weeks administration. In this case dose increases may be
necessary.
Results of clinical studies in children have shown that, on average, the younger the patients, the higher
the NeoRecormon doses
required. Nevertheless, the recommended dosing schedule should be
followed as the individual response cannot be predicted.
3
Treatment with NeoRecormon is normally a
long-term therapy. It can, however, be interrupted, if
necessary, at any time. Data on the once weekly dosing schedule are based on clinical studies with a
treatment duration of 24 weeks.
Treatment of symptomatic chemotherapy-induced anaemia in cancer patients:
NeoRecormon should be administered by the subcutaneous route to patients with anaemia (e.g.
haemoglobin concentration ≤ 10g/dl (6.2 mmol/l)). Anaemia symptoms and sequelae may vary with
age, gender, and overall burden of disease; a physician’s evaluation of the individual patient’s clinical
course and condition is necessary.
The weekly dose can be given as one injection per week or in divided doses 3 to 7 times per week.
The recommended initial dose is 30,000 IU per week (corresponding to approximately 450 IU/kg body
weight per week, based on an average weighted patient).
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.2 mmol/l) to 12 g/dl (7.5 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.5 mmol/l) should be avoided; guidance for appropriate dose adjustment for when haemoglobin
values exceeding 12 g/dl (7.5 mmol/l) are observed are described below.
If, after 4 weeks of therapy, the haemoglobin value has increased by at least 1 g/dl (0.62 mmol/l), the
current dose should be continued. If the haemoglobin value has not increased by at least 1 g/dl
(0.62 mmol/l), a doubling of the weekly dose should be considered. If, after 8 weeks of therapy, the
haemoglobin value has not increased by at least 1 g/dl (0.62 mmol/l), response is unlikely and
treatment should be discontinued.
The therapy should be continued up to 4 weeks after the end of chemotherapy.
The maximum dose should not exceed 60,000 IU per week.
Once the therapeutic objective for an individual patient has been achieved, the dose should be reduced
by 25 to 50 % in order to maintain haemoglobin at that level. Appropriate dose titration should be
considered.
If the haemoglobin exceeds 12 g/dl (7.5 mmol/l), the dose should be reduced by approximately 25 to
50 %. Treatment with NeoRecormon should be temporarily discontinued if haemoglobin levels exceed
13 g/dl (8.1 mmol/l). Therapy should be reinitiated at approximately 25 % lower than the previous
dose after haemoglobin levels fall to 12 g/dl (7.5 mmol/l) or below.
If the rise in haemoglobin is greater than 2 g/dl (1.3 mmol/l) in 4 weeks, the dose should be reduced by
25 to 50 %.
Patients should be monitored closely to ensure that the lowest approved dose of NeoRecormon is used
to provide adequate control of the symptoms of anaemia.
Treatment for increasing the amount of autologous blood:
The reconstituted solution is administered intravenously over approx. 2 minutes or subcutaneously.
NeoRecormon is administered twice weekly over 4 weeks. On those occasions where the patient's
PCV allows blood donation, i.e. PCV ≥ 33 %, NeoRecormon is administered at the end of blood
donation.
During the entire treatment period, a PCV of 48 % should not be exceeded.
The dosage must be determined by the surgical team individually for each patient as a function of the
required amount of pre-donated blood and the endogenous red cell reserve:
1.
4
The required amount of pre-donated blood depends on the anticipated blood loss, use of blood
conserving procedures and the physical condition of the patient.
This amount should be that quantity which is expected to be sufficient to avoid homologous
blood transfusions.
2.
The ability to donate blood depends predominantly on the patient's blood volume and baseline
PCV. Both variables determine the endogenous red cell reserve, which can be calculated
according to the following formula.
Women: blood volume [ml] = 41 [ml/kg] x body weight [kg] + 1200 [ml]
Men:
blood volume [ml] = 44 [ml/kg] x body weight [kg] + 1600 [ml]
(body weight ≥ 45 kg)
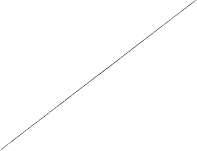
The indication for NeoRecormon treatment and, if given, the single dose should be determined from
the required amount of pre-donated blood and the endogenous red cell reserve according to the
following graphs.
Female patients
Male patients
Required amount of pre-donated blood
Required amount of pre-donated blood
[units]
[units]
Endogenous red cell reserve [ml]
Endogenous red cell reserve [ml]
The single dose thus determined is administered twice weekly over 4 weeks. The maximum dose
should not exceed 1600 IU/kg body weight per week for intravenous or 1200 IU/kg per week for
subcutaneous administration.
Hypersensitivity to the active substance or any of the excipients.
Poorly controlled hypertension.
In the indication "increasing the yield of autologous blood": myocardial infarction or stroke in the
month preceding treatment, unstable angina pectoris, increased risk of deep venous thrombosis such as
history of venous thromboembolic disease.
NeoRecormon Multidose contains benzyl alcohol as a preservative and must therefore not be given to
infants or young children up to three years old.
5
The required amount of pre-donated blood is expressed in units whereby one unit in the
nomogram is equivalent to 180 ml red cells.
Endogenous red cell reserve = blood volume [ml] x (PCV - 33) ÷ 100




































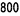





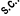















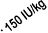







































































NeoRecormon should be used with caution in the presence of refractory anaemia with excess blasts in
transformation, epilepsy, thrombocytosis, and chronic liver failure. Folic acid and vitamin B
12
deficiencies should be ruled out as they reduce the effectiveness of NeoRecormon.
In order to ensure effective erythropoiesis, iron status should be evaluated for all patients prior to and
during treatment and supplementary iron therapy may be necessary and conducted in accordance with
therapeutic guidelines.
Severe aluminium overload due to treatment of renal failure may compromise the effectiveness of
NeoRecormon.
The indication for NeoRecormon treatment of nephrosclerotic patients not yet undergoing dialysis
should be defined individually, as a possible acceleration of progression of renal failure cannot be
ruled out with certainty.
Pure red cell aplasia caused by neutralising anti-erythropoietin antibodies has been reported in
association with erythropoietin therapy, including NeoRecormon. These antibodies have been shown
to cross-react with all erythropoietic proteins, and patients suspected or confirmed to have neutralising
antibodies to erythropoietin should not be switched to NeoRecormon (see section 4.8).
A paradoxical decrease in haemoglobin and development of severe anaemia associated with low
reticulocyte counts should prompt to discontinue treatment with epoetin and perform anti-
erythropoietin antibody testing. Cases have been reported in patients with hepatitis C treated with
interferon and ribavirin, when epoetins are used concomitantly. Epoetins are not approved in the
management of anaemia associated with hepatitis C.
In
chronic renal failure
patients an increase in blood pressure or aggravation of existing hypertension,
especially in cases of rapid PCV increase can occur. These increases in blood pressure can be treated
with medicinal products. If blood pressure rises cannot be controlled by drug therapy, a transient
interruption of NeoRecormon therapy is recommended. Particularly at beginning of therapy, regular
monitoring of the blood pressure is recommended, including between dialyses. Hypertensive crisis
with encephalopathy-like symptoms may occur and require the immediate attention of a physician and
intensive medical care. Particular attention should be paid to sudden stabbing migraine like headaches
as a possible warning sign.
In
chronic renal failure
patients there may be a moderate dose-dependent rise in the platelet count
within the normal range during treatment with NeoRecormon, especially after intravenous
administration. This regresses during the course of continued therapy. It is recommended that the
platelet count be monitored regularly during the first 8 weeks of therapy.
Haemoglobin concentration
In patients with chronic renal failure, maintenance haemoglobin concentration should not exceed the
upper limit of the target haemoglobin concentration recommended in section 4.2. In clinical trials, an
increased risk of death and serious cardiovascular events or cerebrovascular events including stroke
was observed when erythropoiesis stimulating agents (ESAs) were administered to target a
haemoglobin of greater than 12 g/dl (7.5 mmol/l).
Controlled clinical trials have not shown significant benefits attributable to the administration of
epoetins when haemoglobin concentration is increased beyond the level necessary to control
symptoms of anaemia and to avoid blood transfusion.
Effect on tumour growth
Epoetins are growth factors that primarily stimulate red blood cell production. Erythropoietin
receptors may be expressed on the surface of a variety of tumour cells. As with all growth factors,
there is a concern that epoetins could stimulate the growth of tumours. In several controlled studies,
6
epoetins have not been shown to improve overall survival or decrease the risk of tumour progression
in patients with anaemia associated with cancer.
In controlled clinical studies, use of NeoRecormon and other erythropoiesis-stimulating agents (ESAs)
have shown:
-
shortened time to tumour progression in patients with advanced head and neck cancer receiving
radiation therapy when administered to target a haemoglobin of greater than 14 g/dl
(8.7 mmol/l),
-
shortened overall survival and increased deaths attributed to disease progression at 4 months in
patients with metastatic breast cancer receiving chemotherapy when administered to target a
haemoglobin of 12-14 g/dl (7.5-8.7 mmol/l),
-
increased risk of death when administered to target a haemoglobin of 12 g/dl (7.5 mmol/l) in
patients with active malignant disease receiving neither chemotherapy nor radiation therapy.
ESAs are not indicated for use in this patient population.
In view of the above, in some clinical situations blood transfusion should be the preferred treatment
for the management of anaemia in patients with cancer. The decision to administer recombinant
erythropoietins should be based on a benefit-risk assessment with the participation of the individual
patient, which should take into account the specific clinical context. Factors that should be considered
in this assessment should include the type of tumour and its stage; the degree of anaemia; life-
expectancy; the environment in which the patient is being treated; and patient preference (see section
5.1)
There may be an increase in blood pressure which can be treated with drugs. It is therefore
recommended to monitor blood pressure, in particular in the initial treatment phase in cancer patients.
Platelet counts and haemoglobin level should also be monitored at regular intervals in cancer patients.
In patients in an
autologous blood predonation programme
there may be an increase in platelet count,
mostly within the normal range. Therefore, it is recommended that the platelet count be determined at
least once a week in these patients. If there is an increase in platelets of more than 150 x 10
9
/l or if
platelets rise above the normal range, treatment with NeoRecormon should be discontinued.
In chronic renal failure patients an increase in heparin dose during haemodialysis is frequently
required during the course of therapy with NeoRecormon as a result of the increased packed cell
volume. Occlusion of the dialysis system is possible if heparinisation is not optimum.
Early shunt revision and thrombosis prophylaxis by administration of acetylsalicylic acid, for example,
should be considered in chronic renal failure patients at risk of shunt thrombosis.
Serum potassium and phosphate levels should be monitored regularly during NeoRecormon therapy.
Potassium elevation has been reported in a few uraemic patients receiving NeoRecormon, though
causality has not been established. If an elevated or rising potassium level is observed then
consideration should be given to ceasing NeoRecormon administration until the level has been
corrected.
For use of NeoRecormon in an autologous predonation programme, the official guidelines on
principles of blood donation must be considered, in particular:
- only patients with a PCV ≥ 33 % (haemoglobin ≥ 11 g/dl [6.83 mmol/l]) should donate;
- special care should be taken with patients below 50 kg weight;
- the single volume drawn should not exceed approx. 12 % of the patient's estimated blood
volume.
Treatment should be reserved for patients in whom it is considered of particular importance to avoid
homologous blood transfusion taking into consideration the risk/benefit assessment for homologous
transfusions.
Misuse by healthy persons may lead to an excessive increase in packed cell volume. This may be
associated with life-threatening complications of the cardiovascular system.
7
NeoRecormon Multidose contains up to 5.0 mg phenylalanine/vial as an excipient. Therefore this
should be taken into consideration in patients affected with severe forms of phenylketonuria.
NeoRecormon in reconstituted multidose solution contains benzyl alcohol which may cause toxic
reactions and anaphylactoid reactions in infants and children up to 3 years old.
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, i.e. essentially “sodium-
free”.
In order to improve the traceability of ESAs, the name of the prescribed ESA should be clearly
recorded (or: stated) in the patient file.
The clinical results obtained so far do not indicate any interaction of NeoRecormon with other
medicinal products.
Animal experiments revealed that epoetin beta does not increase the myelotoxicity of cytostatic
medicinal products like etoposide, cisplatin, cyclophosphamide, and fluorouracil.
For epoetin beta no clinical data on exposed pregnancies are available. Animal studies do not indicate
direct or indirect harmful effects with respect to pregnancy, embryonal/foetal development, parturition
or postnatal development (see 5.3).
Caution should be exercised when prescribing to pregnant women.
NeoRecormon has no influence on the ability to drive and use machines.
Based on results from clinical trials including 1725 patients approximately 8 % of patients treated with
NeoRecormon are expected to experience adverse reactions.
- Anaemic patients with chronic renal failure
The most frequent adverse reaction during treatment with NeoRecormon is an increase in blood
pressure or aggravation of existing hypertension, especially in cases of rapid PCV increase (see
section 4.4). Hypertensive crisis with encephalopathy-like symptoms (e.g. headaches and confused
state, sensorimotor disorders - such as speech disturbance or impaired gait - up to tonoclonic seizures)
may also occur in individual patients with otherwise
normal or low blood pressure (see section 4.4).
Shunt thromboses may occur, especially in patients who have a tendency to hypotension or
whose arteriovenous fistulae exhibit complications (e.g. stenoses, aneurisms), see section 4.4. In most
cases, a fall in serum ferritin values simultaneous with a rise in packed cell volume is observed (see
section 4.4). In addition, transient increases in serum potassium and phosphate levels have been
observed in isolated cases (see section 4.4).
In isolated cases, neutralising anti erythropoietin antibody-mediated pure red cell aplasia (PRCA)
associated with NeoRecormon therapy has been reported. In case anti-erythropoietin antibody-
mediated PRCA is diagnosed, therapy with NeoRecormon must be discontinued and patients should
not be switched to another erythropoietic protein (see section 4.4).
The incidences of undesirable effects in clinical trials, considered related to treatment with
NeoRecoromon are shown in the table below. Within each frequency grouping, undesirable effects are
presented in order of decreasing seriousness.
8
System Organ Class
Adverse Drug
Reaction
Incidence
Vascular disorders
Hypertensive crisis
Uncommon (>0.1%, <1%)
Hypertension
Common (>1%, <10%)
Nervous system disorders
Headache
Common (>1%, <10%)
Blood and the lymphatic
system disorders
Shunt thrombosis
Thrombocytosis
Rare (>0.01%, <0.1%)
Very rare (<0.01%)
-
Patients with cancer
Epoetin beta treatment-related headache and hypertension which can be treated with drugs are
common (>1 %, <10 %) (see section 4.4).
In some patients, a fall in serum iron parameters is observed (see section 4.4).
Clinical studies have shown a higher frequency of thromboembolic events in cancer patients treated
with NeoRecormon compared to untreated controls or placebo. In patients treated with NeoRecormon,
this incidence is 7 % compared to 4 % in controls; this is not associated with any increase in
thromboembolic mortality compared with controls.
The incidences of undesirable effects in clinical trials, considered related to treatment with
NeoRecoromon are shown in the table below. Within each frequency grouping, undesirable effects are
presented in order of decreasing seriousness.
System Organ Class
Adverse Drug
Reaction
Incidence
Vascular disorders
Hypertension
Common (>1%, <10%)
Blood and the lymphatic
system disorders
Thromboembolic
event
Common (>1%, <10%)
Nervous system disorders
Headache
Common (>1%, <10%)
-
Patients in an autologous blood predonation programme
Patients in an autologous blood predonation programme have been reported to show a slightly higher
frequency of thromboembolic events. However, a causal relationship with treatment with
NeoRecormon could not be established.
In placebo controlled trials temporary iron deficiency was more pronounced in patients treated with
NeoRecormon than in controls (see section 4.4).
The incidences of undesirable effects in clinical trials, considered related to treatment with
NeoRecoromon are shown in the table below. Within each frequency grouping, undesirable effects are
presented in order of decreasing seriousness.
System Organ Class
Adverse Drug
Reaction
Incidence
Nervous system disorders
Headache
Common (>1%, <10%)
-
All indications
Rarely (≥1/10.000 to ≤1/1.000), epoetin beta treatment-related skin reactions such as rash, pruritus,
urticaria or injection site reactions may occur. In very rare cases (≤1/10.000), epoetin beta treatment-
related anaphylactoid reactions have been reported. However, in controlled clinical studies no
increased incidence of hypersensitivity reactions was found.
In very rare cases (≤1/10.000), particularly when starting treatment, epoetin beta treatment-related flu-
like symptoms such as fever, chills, headaches, pain in the limbs, malaise and/or bone pain have been
reported. These reactions were mild or moderate in nature and subsided after a couple of hours or
days.
9
























Data from a controlled clinical trial with epoetin alfa or darbepoetin alfa, reported an incidence of
stroke as common (≥1/100 to <1/10).
The therapeutic margin of NeoRecormon is very wide. Even at very high serum levels no symptoms of
poisoning have been observed.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antianaemic, ATC code: B03XA
Epoetin beta is identical in its amino acid and carbohydrate composition to erythropoietin that has
been isolated from the urine of anaemic patients.
Erythropoietin is a glycoprotein that stimulates the formation of erythrocytes from its committed
progenitors. It acts as a mitosis stimulating factor and differentiation hormone.
The biological efficacy of epoetin beta has been demonstrated after intravenous and subcutaneous
administration in various animal models
in vivo
(normal and uraemic rats, polycythaemic mice, dogs).
After administration of epoetin beta, the number of erythrocytes, the Hb values and reticulocyte counts
increase as well as the
59
Fe-incorporation rate.
An increased
3
H-thymidine incorporation in the erythroid nucleated spleen cells has been found
in
vitro
(mouse spleen cell culture) after incubation with epoetin beta.
Investigations in cell cultures of human bone marrow cells showed that epoetin beta stimulates
erythropoiesis specifically and does not affect leucopoiesis. Cytotoxic actions of epoetin beta on bone
marrow or on human skin cells were not detected.
After single dose administration of epoetin beta no effects on behaviour or locomotor activity of mice
and circulatory or respiratory function of dogs were observed.
In a randomised, double-blind, placebo-controlled study of 4,038 CRF patients not on dialysis with
type 2 diabetes and haemoglobin levels ≤ 11 g/dL, patients received either treatment with darbepoetin
alfa to target haemoglobin levels of 13 g/dL or placebo (see section 4.4). The study did not meet either
primary objective of demonstrating a reduction in risk for all-cause mortality, cardiovascular
morbidity, or end stage renal disease (ESRD). Analysis of the individual components of the composite
endpoints showed the following HR (95% CI): death 1.05 (0.92, 1.21), stroke 1.92 (1.38, 2.68),
congestive heart failure (CHF) 0.89 (0.74, 1.08), myocardial infarction (MI) 0.96 (0.75, 1.23),
hospitalisation for myocardial ischaemia 0.84 (0.55, 1.27), ESRD 1.02 (0.87, 1.18).
Erythropoietin is a growth factor that primarily stimulates red cell production. Erythropoietin receptors
may be expressed on the surface of a variety of tumour cells.
Survival and tumour progression have been examined in five large controlled studies involving a total
of 2833 patients, of which four were double-blind placebo-controlled studies and one was an open-
label study. Two of the studies recruited patients who were being treated with chemotherapy. The
target haemoglobin concentration in two studies was >13 g/dl; in the remaining three studies it was 12-
14 g/dl. In the open-label study there was no difference in overall survival between patients treated
with recombinant human erythropoietin and controls. In the four placebo-controlled studies the hazard
ratios for overall survival ranged between 1.25 and 2.47 in favour of controls. These studies have
shown a consistent unexplained statistically significant excess mortality in patients who have anaemia
associated with various common cancers who received recombinant human erythropoietin compared
to controls. Overall survival outcome in the trials could not be satisfactorily explained by differences
10
in the incidence of thrombosis and related complications between those given recombinant human
erythropoietin and those in the control group.
An individual patient data based meta-analysis, which included data from all 12 controlled clinical
studies in anaemic cancer patients conducted with NeoRecormon (n=2301), showed an overall hazard
ratio point estimate for survival of 1.13 in favour of controls (95 % CI 0.87, 1.46). In patients with
baseline haemoglobin ≤ 10 g/dl (n=899), the hazard ratio point estimate for survival was 0.98 (95 %
CI 0.68 to 1.40). An increased relative risk for thromboembolic events was observed in the overall
population (RR 1.62, 95 % CI: 1.13, 2.31).
A patient-level data analysis has also been performed on more than 13,900 cancer patients (chemo-,
radio-, chemoradio- or no therapy) participating in 53 controlled clinical trials involving several
epoetins. Meta-analysis of overall survival data produced a hazard ratio point estimate of 1.06 in
favour of controls (95% CI: 1.00, 1.12; 53 trials and 13,933 patients) and for cancer patients receiving
chemotherapy, the overall survival hazard ratio was 1.04 (95% CI: 0.97, 1.11; 38 trials and 10, 441
patients). Meta-analyses also indicate consistently a significantly increased relative risk of
thromboembolic events in cancer patients receiving recombinant human erythropoietin (see Section
4.4).
In very rare cases, neutralising anti-erythropoietin antibodies with or without pure red cell aplasia
(PRCA) occurred during rHuEPO therapy.
5.2 Pharmacokinetic properties
Pharmacokinetic investigations in healthy volunteers and uraemic patients show that the half-life of
intravenously administered epoetin beta is between 4 and 12 hours and that the distribution volume
corresponds to one to two times the plasma volume. Analogous results have been found in animal
experiments in uraemic and normal rats.
After subcutaneous administration of epoetin beta to uraemic patients, the protracted absorption results
in a serum concentration plateau, whereby the maximum concentration is reached after an average of
12 - 28 hours. The terminal half-life is higher than after intravenous administration, with an average of
13 - 28 hours.
Bioavailability of epoetin beta after subcutaneous administration is between 23 and 42 % as compared
with intravenous administration.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity, genotoxicity, and toxicity to reproduction.
A carcinogenicity study with homologous erythropoietin in mice did not reveal any signs of
proliferative or tumourigenic potential.
6.
6.1 List of excipients
Lyophilisate:
Urea,
Sodium chloride,
Polysorbate 20,
Sodium dihydrogen phosphate,
Disodium hydrogen phosphate,
Calcium chloride,
Glycine,
11
L-Leucine,
L-Isoleucine,
L-Threonine,
L-Glutamic acid,
L-Phenylalanine.
Solvent:
Benzyl alcohol,
Benzalkonium chloride,
Water for injections.
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in
section 6.6. (content of accompanying solvent ampoule).
6.3 Shelf life
3 years.
Chemical and physical in-use stability of the reconstituted solution has been demonstrated for one
month at 2°C - 8°C. From a microbiological point of view, once opened, the reconstituted solution
may be stored for maximum of one month at 2°C - 8°C. Other in-use storage times and conditions are
the responsibility of the user.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C).
Keep the vial in the outer carton, in order to protect from light.
For the purpose of ambulatory use, the patient may remove the unreconstituted product from the
refrigerator and store it at room temperature (not above 25°C) for one single period of up to 5 days.
Leaving the reconstituted solution outside the refrigerator should be limited to the time necessary for
preparing the injections.
For storage conditions of the reconstituted medicinal product see section 6.3.
6.5 Nature and contents of container
Lyophilisate (50,000 IU) for solution for injection in a vial (type I glass) with a stopper (teflonised
rubber) and 10 ml of solvent in an ampoule (type I glass), with one reconstitution and withdrawal
device with one needle (21G2), and one disposable syringe (polypropylene and polyethylene) (10 ml).
6.6 Special precautions for disposal and other handling
NeoRecormon Multidose is supplied as a lyophilisate for solution for injection in vials. This is
dissolved with the contents of the accompanying solvent ampoule by means of a reconstitution and
withdrawal device according to the instructions given below. Only solutions which are clear or slightly
opalescent, colourless and practically free of visible particles may be injected. Do not use glass
materials for injection, use only plastic materials.
This is a multidose preparation from which different single doses can be withdrawn over a period of
1 month after dissolution. To avoid the risk of contamination of the contents always observe aseptic
techniques (i.e. use disposable sterile syringes and needles to administer each dose) and strictly follow
the handling instructions below. Before withdrawing each dose disinfect the rubber seal of the
withdrawal device with alcohol to prevent contamination of the contents by repeated needle insertions.
12
Preparation of NeoRecormon Multidose solution
(1)
Take the vial with the freeze-dried substance out of the package. Write the date of reconstitution
and expiry on the label (expiry is 1 month after reconstitution).
(2)
Remove the plastic cap from the vial.
(3)
Disinfect the rubber seal with alcohol.
(4)
Take the reconstitution and withdrawal device (which allows sterile air exchange) out of the blister
and remove the protective cover from the spike.
(5)
Attach the device to the vial until the snap lock clicks home.
(6)
Put the green needle on the syringe contained in the package and remove the needle cover.
(7)
Hold the OPC (One-Point-Cut) ampoule with the blue point upwards. Shake or tap the ampoule to
get any fluid in the stem into the body of the ampoule. Take hold of the stem and snap off away from
you. Withdraw
all the solvent
into the syringe. Disinfect the rubber seal of the device with alcohol.
(8)
Penetrate the seal with the needle to a depth of about 1 cm and slowly inject the solvent into the
vial. Then disconnect the syringe (with needle) from the device.
(9)
Swirl the vial gently until the lyophilisate has dissolved. Do not shake. Check that the solution is
clear, colourless and practically free from particles. Put the protective cap on the top of the device.
(10)
Before and after reconstitution NeoRecormon Multidose must be stored at +2° to +8°C
(refrigerator).
Preparation of a single injection
(1)
Before withdrawing each dose disinfect the rubber seal of the device with alcohol.
(2)
Place a 26G needle onto an appropriate single-use syringe (max.1 ml).
(3)
Remove the needle cover and insert the needle through the rubber seal of the device. Withdraw
NeoRecormon solution into the syringe, expel air from the syringe into the vial and adjust the amount
of NeoRecormon solution in the syringe to the dose prescribed. Then disconnect the syringe (with
needle) from the device.
(4)
Replace the needle by a new one (the new needle should have the size which you normally use for
injections).
(5)
Remove the needle cover and carefully expel air from the needle by holding the syringe vertically
and gently pressing the plunger upwards until a bead of liquid appears at the needle tip.
For subcutaneous injection, clean the skin at the site of injection using an alcohol wipe. Form a skin
fold by pinching the skin between the thumb and the forefinger. Hold the syringe near to the needle
and insert the needle into the skin with a quick, firm action. Inject NeoRecormon solution. Withdraw
the needle quickly and apply pressure over the injection site with a dry, sterile pad.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW
United Kingdom
8.
EU/1/97/031/019
13
9.
Date of the first authorisation: 16 July 1997
Date of the last renewal: 16 July 2007
Detailed information on this medicinal product is available on the website of the European Medicines
Agency http://www.ema.europa.eu/.
14
1.
NeoRecormon Multidose 100,000 IU Lyophilisate and solvent for solution for injection
(20,000 IU/ml)
2.
One vial contains 100,000 international units (IU) corresponding to 830 micrograms epoetin beta*
(recombinant human erythropoietin).
One ampoule contains 5 ml solvent (water for injections with benzyl alcohol and benzalkonium
chloride as preservatives).
One ml of reconstituted solution contains 20,000 IU epoetin beta.
* produced in Chinese Hamster Ovary cells (CHO) by recombinant DNA technology
Excipients:
Phenylalanine (up to 5.0 mg/vial)
Sodium (less than 1 mmol per dose)
Benzyl alcohol (up to 20 mg per multidose solvent ampoule)
For a full list of excipients, see section 6.1.
3.
Lyophilisate and solvent for solution for injection.
White lyophilisate and clear, colourless solvent.
4.
-
Treatment of symptomatic anaemia associated with chronic renal failure (CRF)
in adult and
paediatric patients.
-
Treatment of symptomatic anaemia in adult patients with non-myeloid malignancies receiving
chemotherapy.
-
Its use in this indication must be balanced against the reported increased risk of thromboembolic
events. Treatment should only be given to patients with moderate anaemia (Hb 10 - 13 g/dl
[6.21 - 8.07 mmol/l], no iron deficiency) if blood conserving procedures are not available or
insufficient when the scheduled major elective surgery requires a large volume of blood (4 or
more units of blood for females or 5 or more units for males).
Therapy with NeoRecormon should be initiated by physicians experienced in the above mentioned
indications. As anaphylactoid reactions were observed in isolated cases, it is recommended that the
first dose be administered under medical supervision.
This multidose preparation can be used for several patients. To avoid the risk of cross-infection always
follow aseptic techniques and use disposable sterile syringes and needles for each administration.
Please check that only one vial of NeoRecormon Multidose is in use (i.e. reconstituted) at any one
time.
The reconstituted product is a colourless, clear to slightly opalescent solution.
For instructions on reconstitution of the product before administration, see section 6.6.
15
Increasing the yield of autologous blood from patients in a pre-donation programme.
Treatment of symptomatic anaemia in adult and paediatric chronic renal failure patients:
Anaemia symptoms and sequelae may vary with age, gender, and overall burden of disease; a
physician’s evaluation of the individual patient’s clinical course and condition is necessary.
NeoRecormon should be administered either subcutaneously or intravenously in order to increase
haemoglobin to not greater than 12 g/dl (7.5 mmol/l). Subcutaneous use is preferable in patients who
are not receiving haemodialysis to avoid puncture of peripheral veins. In case of intravenous
administration, the solution should be injected over approx. 2 minutes, e.g. in haemodialysis patients
via the arterio-venous fistula at the end of dialysis.
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.2 mmol/l) to 12 g/dl (7.5 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.5 mmol/l) should be avoided; guidance for appropriate dose adjustment for when haemoglobin
values exceeding 12 g/dl (7.5 mmol/l) are observed are described below.
A rise in haemoglobin of greater than 2 g/dl (1.25 mmol/l) over a four week period should be avoided.
If it occurs, appropriate dose adjustment should be made as provided.If the rate of rise in haemoglobin
is greater than 2 g/dl (1.25 mmol/l) in one month or if the haemoglobin level is increasing and
approaching 12 g/dl (7.45 mmol/l), the dose is to be reduced by approximately 25%. If the
haemoglobin level continues to increase, therapy should be interrupted until the hemoglobin level
begins to decrease, at which point therapy should be restarted at a dose approximately 25% below the
previously administered dose.
Patients should be monitored closely to ensure that the lowest approved dose of NeoRecormon is used
to provide adequate control of the symptoms of anaemia.
In the presence of hypertension or existing cardiovascular, cerebrovascular or peripheral vascular
diseases, the weekly increase in Hb and the target Hb should be determined individually taking into
account the clinical picture.
Treatment with NeoRecormon is divided into two stages.
1.
Correction phase
- Subcutaneous administration:
- The initial dosage is 3 x 20 IU/kg body weight per week. The dosage may be increased every
4 weeks by 3 x 20 IU/kg and week if the increase of Hb is not adequate (< 0.25 g/dl per week).
- The weekly dose can also be divided into daily doses.
- Intravenous administration:
The initial dosage is 3 x 40 IU/kg per week. The dosage may be raised after 4 weeks to 80 IU/kg
three times per week - and by further increments of 20 IU/kg if needed, three times per week, at
monthly intervals.
For both routes of administration, the maximum dose should not exceed 720 IU/kg per week.
2.
Maintenance phase
To maintain an Hb of between 10 and 12 g/dl, the dosage is initially reduced to half of the previously
administered amount. Subsequently, the dose is adjusted at intervals of one or two weeks individually
for the patient (maintenance dose).
In the case of subcutaneous administration, the weekly dose can be given as one injection per week or
in divided doses three or seven times per week. Patients who are stable on a once weekly dosing
regimen may be switched to once every two weeks administration. In this case dose increases may be
necessary.
Results of clinical studies in children have shown that, on average, the younger the patients, the higher
the NeoRecormon doses
required. Nevertheless, the recommended dosing schedule should be
followed as the individual response cannot be predicted.
16
Treatment with NeoRecormon is normally a
long-term therapy. It can, however, be interrupted, if
necessary, at any time. Data on the once weekly dosing schedule are based on clinical studies with a
treatment duration of 24 weeks.
Treatment of symptomatic chemotherapy-induced anaemia in cancer patients:
NeoRecormon should be administered by the subcutaneous route to patients with anaemia (e.g.
haemoglobin concentration ≤ 10g/dl (6.2 mmol/l)). Anaemia symptoms and sequelae may vary with
age, gender, and overall burden of disease; a physician’s evaluation of the individual patient’s clinical
course and condition is necessary.
The weekly dose can be given as one injection per week or in divided doses 3 to 7 times per week.
The recommended initial dose is 30,000 IU per week (corresponding to approximately 450 IU/kg body
weight per week, based on an average weighted patient).
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.2 mmol/l) to 12 g/dl (7.5 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.5 mmol/l) should be avoided; guidance for appropriate dose adjustment for when haemoglobin
values exceeding 12 g/dl (7.5 mmol/l) are observed are described below.
If, after 4 weeks of therapy, the haemoglobin value has increased by at least 1 g/dl (0.62 mmol/l), the
current dose should be continued. If the haemoglobin value has not increased by at least 1 g/dl
(0.62 mmol/l), a doubling of the weekly dose should be considered. If, after 8 weeks of therapy, the
haemoglobin value has not increased by at least 1 g/dl (0.62 mmol/l), response is unlikely and
treatment should be discontinued.
The therapy should be continued up to 4 weeks after the end of chemotherapy.
The maximum dose should not exceed 60,000 IU per week.
Once the therapeutic objective for an individual patient has been achieved, the dose should be reduced
by 25 to 50 % in order to maintain haemoglobin at that level. Appropriate dose titration should be
considered.
If the haemoglobin exceeds 12 g/dl (7.5 mmol/l), the dose should be reduced by approximately 25 to
50 %. Treatment with NeoRecormon should be temporarily discontinued if haemoglobin levels exceed
13 g/dl (8.1 mmol/l). Therapy should be reinitiated at approximately 25 % lower than the previous
dose after haemoglobin levels fall to 12 g/dl (7.5 mmol/l) or below.
If the rise in haemoglobin is greater than 2 g/dl (1.3 mmol/l) in 4 weeks, the dose should be reduced by
25 to 50 %.
Patients should be monitored closely to ensure that the lowest approved dose of NeoRecormon is used
to provide adequate control of the symptoms of anaemia.
Treatment for increasing the amount of autologous blood:
The reconstituted solution is administered intravenously over approx. 2 minutes or subcutaneously.
NeoRecormon is administered twice weekly over 4 weeks. On those occasions where the patient's
PCV allows blood donation, i.e. PCV ≥ 33 %, NeoRecormon is administered at the end of blood
donation.
During the entire treatment period, a PCV of 48 % should not be exceeded.
The dosage must be determined by the surgical team individually for each patient as a function of the
required amount of pre-donated blood and the endogenous red cell reserve:
1.
17
The required amount of pre-donated blood depends on the anticipated blood loss, use of blood
conserving procedures and the physical condition of the patient.
This amount should be that quantity which is expected to be sufficient to avoid homologous
blood transfusions.
2.
The ability to donate blood depends predominantly on the patient's blood volume and baseline
PCV. Both variables determine the endogenous red cell reserve, which can be calculated
according to the following formula.
Women: blood volume [ml] = 41 [ml/kg] x body weight [kg] + 1200 [ml]
Men:
blood volume [ml] = 44 [ml/kg] x body weight [kg] + 1600 [ml]
(body weight ≥ 45 kg)
The indication for NeoRecormon treatment and, if given, the single dose should be determined from
the required amount of pre-donated blood and the endogenous red cell reserve according to the
following graphs.
Female patients
Male patients
Required amount of pre-donated blood
Required amount of pre-donated blood
[units]
[units]
Endogenous red cell reserve [ml]
Endogenous red cell reserve [ml]
The single dose thus determined is administered twice weekly over 4 weeks. The maximum dose
should not exceed 1600 IU/kg body weight per week for intravenous or 1200 IU/kg per week for
subcutaneous administration.
Hypersensitivity to the active substance or any of the excipients.
Poorly controlled hypertension.
In the indication "increasing the yield of autologous blood": myocardial infarction or stroke in the
month preceding treatment, unstable angina pectoris, increased risk of deep venous thrombosis such as
history of venous thromboembolic disease.
NeoRecormon Multidose contains benzyl alcohol as a preservative and must therefore not be given to
infants or young children up to three years old.
18
The required amount of pre-donated blood is expressed in units whereby one unit in the
nomogram is equivalent to 180 ml red cells.
Endogenous red cell reserve = blood volume [ml] x (PCV - 33) ÷ 100




































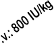











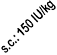



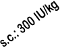











































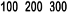
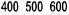






























NeoRecormon should be used with caution in the presence of refractory anaemia with excess blasts in
transformation, epilepsy, thrombocytosis, and chronic liver failure. Folic acid and vitamin B
12
deficiencies should be ruled out as they reduce the effectiveness of NeoRecormon.
In order to ensure effective erythropoiesis, iron status should be evaluated for all patients prior to and
during treatment and supplementary iron therapy may be necessary and conducted in accordance with
therapeutic guidelines.
Severe aluminium overload due to treatment of renal failure may compromise the effectiveness of
NeoRecormon.
The indication for NeoRecormon treatment of nephrosclerotic patients not yet undergoing dialysis
should be defined individually, as a possible acceleration of progression of renal failure cannot be
ruled out with certainty.
Pure red cell aplasia caused by neutralising anti-erythropoietin antibodies has been reported in
association with erythropoietin therapy, including NeoRecormon. These antibodies have been shown
to cross-react with all erythropoietic proteins, and patients suspected or confirmed to have neutralising
antibodies to erythropoietin should not be switched to NeoRecormon (see section 4.8).
A paradoxical decrease in haemoglobin and development of severe anaemia associated with low
reticulocyte counts should prompt to discontinue treatment with epoetin and perform anti-
erythropoietin antibody testing. Cases have been reported in patients with hepatitis C treated with
interferon and ribavirin, when epoetins are used concomitantly. Epoetins are not approved in the
management of anaemia associated with hepatitis C.
In
chronic renal failure
patients an increase in blood pressure or aggravation of existing hypertension,
especially in cases of rapid PCV increase can occur. These increases in blood pressure can be treated
with medicinal products. If blood pressure rises cannot be controlled by drug therapy, a transient
interruption of NeoRecormon therapy is recommended. Particularly at beginning of therapy, regular
monitoring of the blood pressure is recommended, including between dialyses. Hypertensive crisis
with encephalopathy-like symptoms may occur and require the immediate attention of a physician and
intensive medical care. Particular attention should be paid to sudden stabbing migraine like headaches
as a possible warning sign.
In
chronic renal failure
patients there may be a moderate dose-dependent rise in the platelet count
within the normal range during treatment with NeoRecormon, especially after intravenous
administration. This regresses during the course of continued therapy. It is recommended that the
platelet count be monitored regularly during the first 8 weeks of therapy.
Haemoglobin concentration
In patients with chronic renal failure, maintenance haemoglobin concentration should not exceed the
upper limit of the target haemoglobin concentration recommended in section 4.2. In clinical trials, an
increased risk of death and serious cardiovascular events or cerebrovascular events including stroke
was observed when erythropoiesis stimulating agents (ESAs) were administered to target a
haemoglobin of greater than 12 g/dl (7.5 mmol/l).
Controlled clinical trials have not shown significant benefits attributable to the administration of
epoetins when haemoglobin concentration is increased beyond the level necessary to control
symptoms of anaemia and to avoid blood transfusion.
Effect on tumour growth
Epoetins are growth factors that primarily stimulate red blood cell production. Erythropoietin
receptors may be expressed on the surface of a variety of tumour cells. As with all growth factors,
there is a concern that epoetins could stimulate the growth of tumours. In several controlled studies,
19
epoetins have not been shown to improve overall survival or decrease the risk of tumour progression
in patients with anaemia associated with cancer.
In controlled clinical studies, use of NeoRecormon and other erythropoiesis-stimulating agents (ESAs)
have shown:
-
shortened time to tumour progression in patients with advanced head and neck cancer receiving
radiation therapy when administered to target a haemoglobin of greater than 14 g/dl
(8.7 mmol/l),
-
shortened overall survival and increased deaths attributed to disease progression at 4 months in
patients with metastatic breast cancer receiving chemotherapy when administered to target a
haemoglobin of 12-14 g/dl (7.5-8.7 mmol/l),
-
increased risk of death when administered to target a haemoglobin of 12 g/dl (7.5 mmol/l) in
patients with active malignant disease receiving neither chemotherapy nor radiation therapy.
ESAs are not indicated for use in this patient population.
In view of the above, in some clinical situations blood transfusion should be the preferred treatment
for the management of anaemia in patients with cancer. The decision to administer recombinant
erythropoietins should be based on a benefit-risk assessment with the participation of the individual
patient, which should take into account the specific clinical context. Factors that should be considered
in this assessment should include the type of tumour and its stage; the degree of anaemia; life-
expectancy; the environment in which the patient is being treated; and patient preference (see section
5.1)
There may be an increase in blood pressure which can be treated with drugs. It is therefore
recommended to monitor blood pressure, in particular in the initial treatment phase in cancer patients.
Platelet counts and haemoglobin level should also be monitored at regular intervals in cancer patients.
In patients in an
autologous blood predonation programme
there may be an increase in platelet count,
mostly within the normal range. Therefore, it is recommended that the platelet count be determined at
least once a week in these patients. If there is an increase in platelets of more than 150 x 10
9
/l or if
platelets rise above the normal range, treatment with NeoRecormon should be discontinued.
In chronic renal failure patients an increase in heparin dose during haemodialysis is frequently
required during the course of therapy with NeoRecormon as a result of the increased packed cell
volume. Occlusion of the dialysis system is possible if heparinisation is not optimum.
Early shunt revision and thrombosis prophylaxis by administration of acetylsalicylic acid, for example,
should be considered in chronic renal failure patients at risk of shunt thrombosis.
Serum potassium and phosphate levels should be monitored regularly during NeoRecormon therapy.
Potassium elevation has been reported in a few uraemic patients receiving NeoRecormon, though
causality has not been established. If an elevated or rising potassium level is observed then
consideration should be given to ceasing NeoRecormon administration until the level has been
corrected.
For use of NeoRecormon in an autologous predonation programme, the official guidelines on
principles of blood donation must be considered, in particular:
- only patients with a PCV ≥ 33 % (haemoglobin ≥ 11 g/dl [6.83 mmol/l]) should donate;
- special care should be taken with patients below 50 kg weight;
- the single volume drawn should not exceed approx. 12 % of the patient's estimated blood
volume.
Treatment should be reserved for patients in whom it is considered of particular importance to avoid
homologous blood transfusion taking into consideration the risk/benefit assessment for homologous
transfusions.
Misuse by healthy persons may lead to an excessive increase in packed cell volume. This may be
associated with life-threatening complications of the cardiovascular system.
20
NeoRecormon Multidose contains up to 5.0 mg phenylalanine/vial as an excipient. Therefore this
should be taken into consideration in patients affected with severe forms of phenylketonuria.
NeoRecormon in reconstituted multidose solution contains benzyl alcohol which may cause toxic
reactions and anaphylactoid reactions in infants and children up to 3 years old.
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, i.e. essentially “sodium-
free”.
In order to improve the traceability of ESAs, the name of the prescribed ESA should be clearly
recorded (or: stated) in the patient file.
The clinical results obtained so far do not indicate any interaction of NeoRecormon with other
medicinal products.
Animal experiments revealed that epoetin beta does not increase the myelotoxicity of cytostatic
medicinal products like etoposide, cisplatin, cyclophosphamide, and fluorouracil.
For epoetin beta no clinical data on exposed pregnancies are available. Animal studies do not indicate
direct or indirect harmful effects with respect to pregnancy, embryonal/foetal development, parturition
or postnatal development (see 5.3).
Caution should be exercised when prescribing to pregnant women.
NeoRecormon has no influence on the ability to drive and use machines.
Based on results from clinical trials including 1725 patients approximately 8 % of patients treated with
NeoRecormon are expected to experience adverse reactions.
- Anaemic patients with chronic renal failure
The most frequent adverse reaction during treatment with NeoRecormon is an increase in blood
pressure or aggravation of existing hypertension, especially in cases of rapid PCV increase (see
section 4.4). Hypertensive crisis with encephalopathy-like symptoms (e.g. headaches and confused
state, sensorimotor disorders - such as speech disturbance or impaired gait - up to tonoclonic seizures)
may also occur in individual patients with otherwise
normal or low blood pressure (see section 4.4).
Shunt thromboses may occur, especially in patients who have a tendency to hypotension or
whose arteriovenous fistulae exhibit complications (e.g. stenoses, aneurisms), see section 4.4. In most
cases, a fall in serum ferritin values simultaneous with a rise in packed cell volume is observed (see
section 4.4). In addition, transient increases in serum potassium and phosphate levels have been
observed in isolated cases (see section 4.4).
In isolated cases, neutralising anti erythropoietin antibody-mediated pure red cell aplasia (PRCA)
associated with NeoRecormon therapy has been reported. In case anti-erythropoietin antibody-
mediated PRCA is diagnosed, therapy with NeoRecormon must be discontinued and patients should
not be switched to another erythropoietic protein (see section 4.4).
The incidences of undesirable effects in clinical trials, considered related to treatment with
NeoRecoromon are shown in the table below. Within each frequency grouping, undesirable effects are
presented in order of decreasing seriousness.
21
System Organ Class
Adverse Drug
Reaction
Incidence
Vascular disorders
Hypertensive crisis
Uncommon (>0.1%, <1%)
Hypertension
Common (>1%, <10%)
Nervous system disorders
Headache
Common (>1%, <10%)
Blood and the lymphatic
system disorders
Shunt thrombosis
Thrombocytosis
Rare (>0.01%, <0.1%)
Very rare (<0.01%)
-
Patients with cancer
Epoetin beta treatment-related headache and hypertension which can be treated with drugs are
common (>1%, <10%) (see section 4.4).
In some patients, a fall in serum iron parameters is observed (see section 4.4).
Clinical studies have shown a higher frequency of thromboembolic events in cancer patients treated
with NeoRecormon compared to untreated controls or placebo. In patients treated with NeoRecormon,
this incidence is 7 % compared to 4 % in controls; this is not associated with any increase in
thromboembolic mortality compared with controls.
The incidences of undesirable effects in clinical trials, considered related to treatment with
NeoRecoromon are shown in the table below. Within each frequency grouping, undesirable effects are
presented in order of decreasing seriousness.
System Organ Class
Adverse Drug
Reaction
Incidence
Vascular disorders
Hypertension
Common (>1%, <10%)
Blood and the lymphatic
system disorders
Thromboembolic
event
Common (>1%, <10%)
Nervous system disorders
Headache
Common (>1%, <10%)
-
Patients in an autologous blood predonation programme
Patients in an autologous blood predonation programme have been reported to show a slightly higher
frequency of thromboembolic events. However, a causal relationship with treatment with
NeoRecormon could not be established.
In placebo controlled trials temporary iron deficiency was more pronounced in patients treated with
NeoRecormon than in controls (see section 4.4).
The incidences of undesirable effects in clinical trials, considered related to treatment with
NeoRecoromon are shown in the table below. Within each frequency grouping, undesirable effects are
presented in order of decreasing seriousness.
System Organ Class
Adverse Drug
Reaction
Incidence
Nervous system disorders
Headache
Common (>1%, <10%)
-
All indications
Rarely (≥1/10.000 to ≤1/1.000), epoetin beta treatment-related skin reactions such as rash, pruritus,
urticaria or injection site reactions may occur. In very rare cases (≤1/10.000), epoetin beta treatment-
related anaphylactoid reactions have been reported. However, in controlled clinical studies no
increased incidence of hypersensitivity reactions was found.
In very rare cases (≤1/10.000), particularly when starting treatment, epoetin beta treatment-related flu-
like symptoms such as fever, chills, headaches, pain in the limbs, malaise and/or bone pain have been
reported. These reactions were mild or moderate in nature and subsided after a couple of hours or
days.
22
























Data from a controlled clinical trial with epoetin alfa or darbepoetin alfa, reported an incidence of
stroke as common (≥1/100 to <1/10).
The therapeutic margin of NeoRecormon is very wide. Even at very high serum levels no symptoms of
poisoning have been observed.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antianaemic, ATC code: B03XA
Epoetin beta is identical in its amino acid and carbohydrate composition to erythropoietin that has
been isolated from the urine of anaemic patients.
Erythropoietin is a glycoprotein that stimulates the formation of erythrocytes from its committed
progenitors. It acts as a mitosis stimulating factor and differentiation hormone.
The biological efficacy of epoetin beta has been demonstrated after intravenous and subcutaneous
administration in various animal models
in vivo
(normal and uraemic rats, polycythaemic mice, dogs).
After administration of epoetin beta, the number of erythrocytes, the Hb values and reticulocyte counts
increase as well as the
59
Fe-incorporation rate.
An increased
3
H-thymidine incorporation in the erythroid nucleated spleen cells has been found
in
vitro
(mouse spleen cell culture) after incubation with epoetin beta.
Investigations in cell cultures of human bone marrow cells showed that epoetin beta stimulates
erythropoiesis specifically and does not affect leucopoiesis. Cytotoxic actions of epoetin beta on bone
marrow or on human skin cells were not detected.
After single dose administration of epoetin beta no effects on behaviour or locomotor activity of mice
and circulatory or respiratory function of dogs were observed.
In a randomised, double-blind, placebo-controlled study of 4,038 CRF patients not on dialysis with
type 2 diabetes and haemoglobin levels ≤ 11 g/dL, patients received either treatment with darbepoetin
alfa to target haemoglobin levels of 13 g/dL or placebo (see section 4.4). The study did not meet either
primary objective of demonstrating a reduction in risk for all-cause mortality, cardiovascular
morbidity, or end stage renal disease (ESRD). Analysis of the individual components of the composite
endpoints showed the following HR (95% CI): death 1.05 (0.92, 1.21), stroke 1.92 (1.38, 2.68),
congestive heart failure (CHF) 0.89 (0.74, 1.08), myocardial infarction (MI) 0.96 (0.75, 1.23),
hospitalisation for myocardial ischaemia 0.84 (0.55, 1.27), ESRD 1.02 (0.87, 1.18).
Erythropoietin is a growth factor that primarily stimulates red cell production. Erythropoietin receptors
may be expressed on the surface of a variety of tumour cells.
Survival and tumour progression have been examined in five large controlled studies involving a total
of 2833 patients, of which four were double-blind placebo-controlled studies and one was an open-
label study. Two of the studies recruited patients who were being treated with chemotherapy. The
target haemoglobin concentration in two studies was >13 g/dl; in the remaining three studies it was 12-
14 g/dl. In the open-label study there was no difference in overall survival between patients treated
with recombinant human erythropoietin and controls. In the four placebo-controlled studies the hazard
ratios for overall survival ranged between 1.25 and 2.47 in favour of controls. These studies have
shown a consistent unexplained statistically significant excess mortality in patients who have anaemia
associated with various common cancers who received recombinant human erythropoietin compared
to controls. Overall survival outcome in the trials could not be satisfactorily explained by differences
23
in the incidence of thrombosis and related complications between those given recombinant human
erythropoietin and those in the control group.
An individual patient data based meta-analysis, which included data from all 12 controlled clinical
studies in anaemic cancer patients conducted with NeoRecormon (n=2301), showed an overall hazard
ratio point estimate for survival of 1.13 in favour of controls (95 % CI 0.87, 1.46). In patients with
baseline haemoglobin ≤ 10 g/dl (n=899), the hazard ratio point estimate for survival was 0.98 (95 %
CI 0.68 to 1.40). An increased relative risk for thromboembolic events was observed in the overall
population (RR 1.62, 95% CI: 1.13, 2.31).
A patient-level data analysis has also been performed on more than 13,900 cancer patients (chemo-,
radio-, chemoradio- or no therapy) participating in 53 controlled clinical trials involving several
epoetins. Meta-analysis of overall survival data produced a hazard ratio point estimate of 1.06 in
favour of controls (95% CI: 1.00, 1.12; 53 trials and 13,933 patients) and for cancer patients receiving
chemotherapy, the overall survival hazard ratio was 1.04 (95% CI: 0.97, 1.11; 38 trials and 10, 441
patients). Meta-analyses also indicate consistently a significantly increased relative risk of
thromboembolic events in cancer patients receiving recombinant human erythropoietin (see Section
4.4).
In very rare cases, neutralising anti-erythropoietin antibodies with or without pure red cell aplasia
(PRCA) occurred during rHuEPO therapy.
5.2 Pharmacokinetic properties
Pharmacokinetic investigations in healthy volunteers and uraemic patients show that the half-life of
intravenously administered epoetin beta is between 4 and 12 hours and that the distribution volume
corresponds to one to two times the plasma volume. Analogous results have been found in animal
experiments in uraemic and normal rats.
After subcutaneous administration of epoetin beta to uraemic patients, the protracted absorption results
in a serum concentration plateau, whereby the maximum concentration is reached after an average of
12 - 28 hours. The terminal half-life is higher than after intravenous administration, with an average of
13 - 28 hours.
Bioavailability of epoetin beta after subcutaneous administration is between 23 and 42 % as compared
with intravenous administration.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity, genotoxicity, and toxicity to reproduction.
A carcinogenicity study with homologous erythropoietin in mice did not reveal any signs of
proliferative or tumourigenic potential.
6.
6.1 List of excipients
Lyophilisate:
Urea,
Sodium chloride,
Polysorbate 20,
Sodium dihydrogen phosphate,
Disodium hydrogen phosphate,
Calcium chloride,
Glycine,
24
L-Leucine,
L-Isoleucine,
L-Threonine,
L-Glutamic acid,
L-Phenylalanine.
Solvent:
Benzyl alcohol,
Benzalkonium chloride,
Water for injections
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in
section 6.6. (content of accompanying solvent ampoule).
6.3 Shelf life
3 years.
Chemical and physical in-use stability of the reconstituted solution has been demonstrated for one
month at 2°C-8°C. From a microbiological point of view, once opened, the reconstituted solution may
be stored for maximum of one month at 2°C-8°C. Other in-use storage times and conditions are the
responsibility of the user.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C).
Keep the vial in the outer carton, in order to protect from light.
For the purpose of ambulatory use, the patient may remove the unreconstituted product from the
refrigerator and store it at room temperature (not above 25°C) for one single period of up to 5 days.
Leaving the reconstituted solution outside the refrigerator should be limited to the time necessary for
preparing the injections.
For storage conditions of the reconstituted medicinal product see section 6.3.
6.5 Nature and contents of container
Lyophilisate (100,000 IU) for solution for injection in a vial (type 1 glass) with a stopper (teflonised
rubber) and 5 ml of solvent in an ampoule (type 1 glass), with one reconstitution and withdrawal
device with one needle (21G2) and one disposable syringe (polypropylene and polyethylene) (5 ml).
6.6 Special precautions for disposal and other handling
NeoRecormon Multidose is supplied as a lyophilisate for solution for injection in vials. This is
dissolved with the contents of the accompanying solvent ampoule by means of a reconstitution and
withdrawal device according to the instructions given below. Only solutions which are clear or slightly
opalescent, colourless and practically free of visible particles may be injected. Do not use glass
materials for injection, use only plastic materials.
This is a multidose preparation from which different single doses can be withdrawn over a period of
1 month after dissolution. To avoid the risk of contamination of the contents always observe aseptic
techniques (i.e. use disposable sterile syringes and needles to administer each dose) and strictly follow
the handling instructions below. Before withdrawing each dose disinfect the rubber seal of the
withdrawal device with alcohol to prevent contamination of the contents by repeated needle insertions.
25
Preparation of NeoRecormon Multidose solution
(1)
Take the vial with the freeze-dried substance out of the package. Write the date of reconstitution
and expiry on the label (expiry is 1 month after reconstitution).
(2)
Remove the plastic cap from the vial.
(3)
Disinfect the rubber seal with alcohol.
(4)
Take the reconstitution and withdrawal device (which allows sterile air exchange) out of the blister
and remove the protective cover from the spike.
(5)
Attach the device to the vial until the snap lock clicks home.
(6)
Put the green needle on the syringe contained in the package and remove the needle cover.
(7)
Hold the OPC (One-Point-Cut) ampoule with the blue point upwards. Shake or tap the ampoule to
get any fluid in the stem into the body of the ampoule. Take hold of the stem and snap off away from
you. Withdraw
all the solvent
into the syringe. Disinfect the rubber seal of the device with alcohol.
(8)
Penetrate the seal with the needle to a depth of about 1 cm and slowly inject the solvent into the
vial. Then disconnect the syringe (with needle) from the device.
(9)
Swirl the vial gently until the lyophilisate has dissolved. Do not shake. Check that the solution is
clear, colourless and practically free from particles. Put the protective cap on the top of the device.
(10)
Before and after reconstitution NeoRecormon Multidose must be stored at +2° to +8°C
(refrigerator).
Preparation of a single injection
(1)
Before withdrawing each dose disinfect the rubber seal of the device with alcohol.
(2)
Place a 26G needle onto an appropriate single-use syringe (max.1 ml).
(3)
Remove the needle cover and insert the needle through the rubber seal of the device. Withdraw
NeoRecormon solution into the syringe, expel air from the syringe into the vial and adjust the amount
of NeoRecormon solution in the syringe to the dose prescribed. Then disconnect the syringe (with
needle) from the device.
(4)
Replace the needle by a new one (the new needle should have the size which you normally use for
injections).
(5)
Remove the needle cover and carefully expel air from the needle by holding the syringe vertically
and gently pressing the plunger upwards until a bead of liquid appears at the needle tip.
For subcutaneous injection, clean the skin at the site of injection using an alcohol wipe. Form a skin
fold by pinching the skin between the thumb and the forefinger. Hold the syringe near to the needle
and insert the needle into the skin with a quick, firm action. Inject NeoRecormon solution. Withdraw
the needle quickly and apply pressure over the injection site with a dry, sterile pad.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW
United Kingdom
8.
EU/1/97/031/020
26
9.
Date of the first authorisation: 16 July 1997
Date of the last renewal: 16 July 2007
Detailed information on this medicinal product is available on the website of the European Medicines
Agency http://www.emea.europa.eu/.
27
1.
NeoRecormon 10,000 IU Lyophilisate and solvent for solution for injection in cartridge
(10,000 IU/ml)
2.
One cartridge contains 10,000 international units (IU) corresponding to 83 micrograms epoetin beta*
(recombinant human erythropoietin) and 1
ml solvent (water for injections with benzyl alcohol and
benzalkonium chloride as preservatives).
One ml solution for injection contains 10,000 IU epoetin beta.
* produced in Chinese Hamster Ovary cells (CHO) by recombinant DNA technology
Excipients:
Phenylalanine (up to 0.5 mg/cartridge)
Sodium (less than 1 mmol/cartridge)
Benzyl alcohol (up to 4 mg/cartridge)
For a full list of excipients, see section 6.1.
3.
Lyophilisate and solvent for solution for injection.
White lyophilisate and clear, colourless solvent.
4.
-
Treatment of symptomatic anaemia associated with chronic renal failure (CRF)
in adult and
paediatric patients.
-
Treatment of symptomatic anaemia in adult patients with non-myeloid malignancies receiving
chemotherapy.
-
Increasing the yield of autologous blood from patients in a pre-donation programme.
Its use in this indication must be balanced against the reported increased risk of thromboembolic
events. Treatment should only be given to patients with moderate anaemia (Hb 10 - 13 g/dl
[6.21 - 8.07 mmol/l], no iron deficiency) if blood conserving procedures are not available or
insufficient when the scheduled major elective surgery requires a large volume of blood (4 or
more units of blood for females or 5 or more units for males).
Therapy with NeoRecormon should be initiated by physicians experienced in the above mentioned
indications. As anaphylactoid reactions were observed in isolated cases, it is recommended that the
first dose be administered under medical supervision.
The reconstituted product is a colourless, clear to slightly opalescent solution.
For instructions on reconstitution of the product before administration, see section 6.6.
The prepared solution is administered subcutaneously.
28
Treatment of symptomatic anaemia in adult and paediatric chronic renal failure patients:
Anaemia symptoms and sequelae may vary with age, gender, and overall burden of disease; a
physician’s evaluation of the individual patient’s clinical course and condition is necessary.
NeoRecormon should be administered subcutaneously in order to increase haemoglobin to not greater
than 12 g/dl (7.5 mmol/l).
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.2 mmol/l) to 12 g/dl (7.5 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.5 mmol/l) should be avoided; guidance for appropriate dose adjustment for when haemoglobin
values exceeding 12 g/dl (7.5 mmol/l) are observed are described below.
A rise in haemoglobin of greater than 2 g/dl (1.25 mmol/l) over a four week period should be avoided.
If it occurs, appropriate dose adjustment should be made as provided. If the rate of rise in haemoglobin
is greater than 2 g/dl (1.25 mmol/l) in one month or if the haemoglobin level is increasing and
approaching 12 g/dl (7.45 mmol/l), the dose is to be reduced by approximately 25 %. If the
haemoglobin level continues to increase, therapy should be interrupted until the hemoglobin level
begins to decrease, at which point therapy should be restarted at a dose approximately 25 % below the
previously administered dose.
Patients should be monitored closely to ensure that the lowest approved dose of NeoRecormon is used
to provide adequate control of the symptoms of anaemia.
In the presence of hypertension or existing cardiovascular, cerebrovascular or peripheral vascular
diseases, the weekly increase in Hb and the target Hb should be determined individually taking into
account the clinical picture.
Treatment with NeoRecormon is divided into two stages.
1.
Correction phase
The initial dosage is 3 x 20 IU/kg body weight per week. The dosage may be increased every 4 weeks
by 3 x 20 IU/kg and week if the increase of Hb is not adequate (< 0.25 g/dl per week).
The weekly dose can also be divided into daily doses.
The maximum dose should not exceed 720 IU/kg per week.
2.
Maintenance phase
To maintain an Hb of between 10 and 12 g/dl, the dosage is initially reduced to half of the previously
administered amount. Subsequently, the dose is adjusted at intervals of one or two weeks individually
for the patient (maintenance dose).
The weekly dose can be given as one injection per week or in divided doses three or seven times per
week. Patients who are stable on a once weekly dosing regimen may be switched to once every two
weeks administration. In this case dose increases may be necessary.
Results of clinical studies in children have shown that, on average, the younger the patients, the higher
the NeoRecormon doses
required. Nevertheless, the recommended dosing schedule should be
followed as the individual response cannot be predicted.
Treatment with NeoRecormon is normally a
long-term therapy. It can, however, be interrupted, if
necessary, at any time. Data on the once weekly dosing schedule are based on clinical studies with a
treatment duration of 24 weeks.
Treatment of symptomatic chemotherapy-induced anaemia in cancer patients:
NeoRecormon should be administered by the subcutaneous route to patients with anaemia (e.g.
haemoglobin concentration ≤ 10g/dl (6.2 mmol/l)). Anaemia symptoms and sequelae may vary with
age, gender, and overall burden of disease; a physician’s evaluation of the individual patient’s clinical
course and condition is necessary.
29
The weekly dose can be given as one injection per week or in divided doses 3 to 7 times per week.
The recommended initial dose is 30,000 IU per week (corresponding to approximately 450 IU/kg body
weight per week, based on an average weighted patient).
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management, with consideration for the haemoglobin target range of 10 g/dl
(6.2 mmol/l) to 12 g/dl (7.5 mmol/l). A sustained haemoglobin level of greater than 12 g/dl
(7.5 mmol/l) should be avoided; guidance for appropriate dose adjustment for when haemoglobin
values exceeding 12 g/dl (7.5 mmol/l) are observed are described below.
If, after 4 weeks of therapy, the haemoglobin value has increased by at least 1 g/dl (0.62 mmol/l), the
current dose should be continued. If the haemoglobin value has not increased by at least 1 g/dl
(0.62 mmol/l), a doubling of the weekly dose should be considered. If, after 8 weeks of therapy, the
haemoglobin value has not increased by at least 1 g/dl (0.62 mmol/l), response is unlikely and
treatment should be discontinued.
The therapy should be continued up to 4 weeks after the end of chemotherapy.
The maximum dose should not exceed 60,000 IU per week.
Once the therapeutic objective for an individual patient has been achieved, the dose should be reduced
by 25 to 50 % in order to maintain haemoglobin at that level. Appropriate dose titration should be
considered.
If the haemoglobin exceeds 12 g/dl (7.5 mmol/l), the dose should be reduced by approximately 25 to
50 %. Treatment with NeoRecormon should be temporarily discontinued if haemoglobin levels exceed
13 g/dl (8.1 mmol/l). Therapy should be reinitiated at approximately 25 % lower than the previous
dose after haemoglobin levels fall to 12 g/dl (7.5 mmol/l) or below.
If the rise in haemoglobin is greater than 2 g/dl (1.3 mmol/l) in 4 weeks, the dose should be reduced by
25 to 50 %.
Patients should be monitored closely to ensure that the lowest approved dose of NeoRecormon is used
to provide adequate control of the symptoms of anaemia.
Treatment for increasing the amount of autologous blood:
NeoRecormon is administered twice weekly over 4 weeks. On those occasions where the patient's
PCV allows blood donation, i.e. PCV ≥ 33 %, NeoRecormon is administered at the end of blood
donation.
During the entire treatment period, a PCV of 48 % should not be exceeded.
The dosage must be determined by the surgical team individually for each patient as a function of the
required amount of pre-donated blood and the endogenous red cell reserve:
1.
The required amount of pre-donated blood depends on the anticipated blood loss, use of blood
conserving procedures and the physical condition of the patient.
This amount should be that quantity which is expected to be sufficient to avoid homologous
blood transfusions.
The required amount of pre-donated blood is expressed in units whereby one unit in the
nomogram is equivalent to 180 ml red cells.
2.
The ability to donate blood depends predominantly on the patient's blood volume and baseline
PCV. Both variables determine the endogenous red cell reserve, which can be calculated
according to the following formula.
30
Women: blood volume [ml] = 41 [ml/kg] x body weight [kg] + 1200 [ml]
Men:
blood volume [ml] = 44 [ml/kg] x body weight [kg] + 1600 [ml]
(body weight ≥ 45 kg)
The indication for NeoRecormon treatment and, if given, the single dose should be determined from
the required amount of pre-donated blood and the endogenous red cell reserve according to the
following graphs.
Female patients
Male patients
Required amount of pre-donated blood
Required amount of pre-donated blood
[units]
[units]
Endogenous red cell reserve [ml]
Endogenous red cell reserve [ml]
The single dose thus determined is administered twice weekly over 4 weeks. The maximum dose
should not exceed 1200 IU/kg body weight per week.
Hypersensitivity to the active substance or any of the excipients.
Poorly controlled hypertension.
In the indication "increasing the yield of autologous blood": myocardial infarction or stroke in the
month preceding treatment, unstable angina pectoris, increased risk of deep venous thrombosis such as
history of venous thromboembolic disease.
NeoRecormon in cartridge contains benzyl alcohol as a preservative and must therefore not be given
to infants or young children up to three years old.
NeoRecormon should be used with caution in the presence of refractory anaemia with excess blasts in
transformation, epilepsy, thrombocytosis, and chronic liver failure. Folic acid and vitamin B
12
deficiencies should be ruled out as they reduce the effectiveness of NeoRecormon.
In order to ensure effective erythropoiesis, iron status should be evaluated for all patients prior to and
during treatment and supplementary iron therapy may be necessary and conducted in accordance with
therapeutic guidelines.
31
Endogenous red cell reserve = blood volume [ml] x (PCV - 33) ÷ 100




























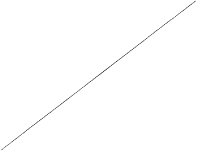






















































































Severe aluminium overload due to treatment of renal failure may compromise the effectiveness of
NeoRecormon.
The indication for NeoRecormon treatment of nephrosclerotic patients not yet undergoing dialysis
should be defined individually, as a possible acceleration of progression of renal failure cannot be
ruled out with certainty.
Pure red cell aplasia caused by neutralising anti-erythropoietin antibodies has been reported in
association with erythropoietin therapy, including NeoRecormon. These antibodies have been shown
to cross-react with all erythropoietic proteins, and patients suspected or confirmed to have neutralising
antibodies to erythropoietin should not be switched to NeoRecormon (see section 4.8).
A paradoxical decrease in haemoglobin and development of severe anaemia associated with low
reticulocyte counts should prompt to discontinue treatment with epoetin and perform anti-
erythropoietin antibody testing. Cases have been reported in patients with hepatitis C treated with
interferon and ribavirin, when epoetins are used concomitantly. Epoetins are not approved in the
management of anaemia associated with hepatitis C.
In
chronic renal failure
patients an increase in blood pressure or aggravation of existing hypertension,
especially in cases of rapid PCV increase can occur. These increases in blood pressure can be treated
with medicinal products. If blood pressure rises cannot be controlled by drug therapy, a transient
interruption of NeoRecormon therapy is recommended. Particularly at beginning of therapy, regular
monitoring of the blood pressure is recommended, including between dialyses. Hypertensive crisis
with encephalopathy-like symptoms may occur and require the immediate attention of a physician and
intensive medical care. Particular attention should be paid to sudden stabbing migraine like headaches
as a possible warning sign.
In
chronic renal failure
patients there may be a moderate dose-dependent rise in the platelet count
within the normal range during treatment with NeoRecormon, especially after intravenous
administration. This regresses during the course of continued therapy. It is recommended that the
platelet count be monitored regularly during the first 8 weeks of therapy.
Haemoglobin concentration
In patients with chronic renal failure, maintenance haemoglobin concentration should not exceed the
upper limit of the target haemoglobin concentration recommended in section 4.2. In clinical trials, an
increased risk of death and serious cardiovascular events or cerebrovascular events including stroke
was observed when erythropoiesis stimulating agents (ESAs) were administered to target a
haemoglobin of greater than 12 g/dl (7.5 mmol/l).
Controlled clinical trials have not shown significant benefits attributable to the administration of
epoetins when haemoglobin concentration is increased beyond the level necessary to control
symptoms of anaemia and to avoid blood transfusion.
Effect on tumour growth
Epoetins are growth factors that primarily stimulate red blood cell production. Erythropoietin
receptors may be expressed on the surface of a variety of tumour cells. As with all growth factors,
there is a concern that epoetins could stimulate the growth of tumours. In several controlled studies,
epoetins have not been shown to improve overall survival or decrease the risk of tumour progression
in patients with anaemia associated with cancer.
In controlled clinical studies, use of NeoRecormon and other erythropoiesis-stimulating agents (ESAs)
have shown:
-
shortened time to tumour progression in patients with advanced head and neck cancer receiving
radiation therapy when administered to target a haemoglobin of greater than 14 g/dl
(8.7 mmol/l),
-
shortened overall survival and increased deaths attributed to disease progression at 4 months in
patients with metastatic breast cancer receiving chemotherapy when administered to target a
haemoglobin of 12-14 g/dl (7.5-8.7 mmol/l),
32
-
increased risk of death when administered to target a haemoglobin of 12 g/dl (7.5 mmol/l) in
patients with active malignant disease receiving neither chemotherapy nor radiation therapy.
ESAs are not indicated for use in this patient population.
In view of the above, in some clinical situations blood transfusion should be the preferred treatment
for the management of anaemia in patients with cancer. The decision to administer recombinant
erythropoietins should be based on a benefit-risk assessment with the participation of the individual
patient, which should take into account the specific clinical context. Factors that should be considered
in this assessment should include the type of tumour and its stage; the degree of anaemia; life-
expectancy; the environment in which the patient is being treated; and patient preference (see section
5.1)
There may be an increase in blood pressure which can be treated with drugs. It is therefore
recommended to monitor blood pressure, in particular in the initial treatment phase in cancer patients.
Platelet counts and haemoglobin level should also be monitored at regular intervals in cancer patients.
In patients in an
autologous blood predonation programme
there may be an increase in platelet count,
mostly within the normal range. Therefore, it is recommended that the platelet count be determined at
least once a week in these patients. If there is an increase in platelets of more than 150 x 10
9
/l or if
platelets rise above the normal range, treatment with NeoRecormon should be discontinued.
In chronic renal failure patients an increase in heparin dose during haemodialysis is frequently
required during the course of therapy with NeoRecormon as a result of the increased packed cell
volume. Occlusion of the dialysis system is possible if heparinisation is not optimum.
Early shunt revision and thrombosis prophylaxis by administration of acetylsalicylic acid, for example,
should be considered in chronic renal failure patients at risk of shunt thrombosis.
Serum potassium and phosphate levels should be monitored regularly during NeoRecormon therapy.
Potassium elevation has been reported in a few uraemic patients receiving NeoRecormon, though
causality has not been established. If an elevated or rising potassium level is observed then
consideration should be given to ceasing NeoRecormon administration until the level has been
corrected.
For use of NeoRecormon in an autologous predonation programme, the official guidelines on
principles of blood donation must be considered, in particular:
- only patients with a PCV ≥ 33 % (haemoglobin ≥ 11 g/dl [6.83 mmol/l]) should donate;
- special care should be taken with patients below 50 kg weight;
- the single volume drawn should not exceed approx. 12 % of the patient's estimated blood
volume.
Treatment should be reserved for patients in whom it is considered of particular importance to avoid
homologous blood transfusion taking into consideration the risk/benefit assessment for homologous
transfusions.
Misuse by healthy persons may lead to an excessive increase in packed cell volume. This may be
associated with life-threatening complications of the cardiovascular system.
NeoRecormon in cartridge contains up to 0.5 mg phenylalanine/cartridge as an excipient. Therefore
this should be taken into consideration in patients affected with severe forms of phenylketonuria.
NeoRecormon in cartridge contains up to 4 mg benzyl alcohol which may cause toxic reactions and
anaphylactoid reactions in infants and children up to 3 years old.
This medicinal product contains less than 1 mmol sodium (23 mg) per cartridge, i.e. essentially
“sodium-free”.
33
In order to improve the traceability of ESAs, the name of the prescribed ESA should be clearly
recorded (or: stated) in the patient file.
The clinical results obtained so far do not indicate any interaction of NeoRecormon with other
medicinal products.
Animal experiments revealed that epoetin beta does not increase the myelotoxicity of cytostatic
medicinal products like etoposide, cisplatin, cyclophosphamide, and fluorouracil.
For epoetin beta no clinical data on exposed pregnancies are available. Animal studies do not indicate
direct or indirect harmful effects with respect to pregnancy, embryonal/foetal development, parturition
or postnatal development (see 5.3).
Caution should be exercised when prescribing to pregnant women.
NeoRecormon has no influence on the ability to drive and use machines.
Based on results from clinical trials including 1725 patients approximately 8 % of patients treated with
NeoRecormon are expected to experience adverse reactions.
-
Anaemic patients with chronic renal failure
The most frequent adverse reaction during treatment with NeoRecormon is an increase in blood
pressure or aggravation of existing hypertension, especially in cases of rapid PCV increase (see
section 4.4). Hypertensive crisis with encephalopathy-like symptoms (e.g. headaches and confused
state, sensorimotor disorders - such as speech disturbance or impaired gait - up to tonoclonic seizures)
may also occur in individual patients with otherwise
normal or low blood pressure (see section 4.4).
Shunt thromboses may occur, especially in patients who have a tendency to hypotension or
whose arteriovenous fistulae exhibit complications (e.g. stenoses, aneurisms), see section 4.4. In most
cases, a fall in serum ferritin values simultaneous with a rise in packed cell volume is observed (see
section 4.4). In addition, transient increases in serum potassium and phosphate levels have been
observed in isolated cases (see section 4.4).
In isolated cases, neutralising anti erythropoietin antibody-mediated pure red cell aplasia (PRCA)
associated with NeoRecormon therapy has been reported. In case anti-erythropoietin antibody-
mediated PRCA is diagnosed, therapy with NeoRecormon must be discontinued and patients should
not be switched to another erythropoietic protein (see section 4.4).
The incidences of undesirable effects in clinical trials, considered related to treatment with
NeoRecoromon are shown in the table below. Within each frequency grouping, undesirable effects are
presented in order of decreasing seriousness.
System Organ Class
Adverse Drug
Reaction
Incidence
Vascular disorders
Hypertensive crisis
Uncommon (>0.1%, <1%)
Hypertension
Common (>1%, <10%)
Nervous system disorders
Headache
Common (>1%, <10%)
Blood and the lymphatic
system disorders
Shunt thrombosis
Thrombocytosis
Rare (>0.01%, <0.1%)
Very rare (<0.01%)
34








-
Patients with cancer
Epoetin beta treatment-related headache and hypertension which can be treated with drugs are
common (>1 %, <10 %) (see section 4.4).
In some patients, a fall in serum iron parameters is observed (see section 4.4).
Clinical studies have shown a higher frequency of thromboembolic events in cancer patients treated
with NeoRecormon compared to untreated controls or placebo. In patients treated with NeoRecormon,
this incidence is 7 % compared to 4 % in controls; this is not associated with any increase in
thromboembolic mortality compared with controls.
The incidences of undesirable effects in clinical trials, considered related to treatment with
NeoRecoromon are shown in the table below. Within each frequency grouping, undesirable effects are
presented in order of decreasing seriousness.
System Organ Class
Adverse Drug
Reaction
Incidence
Vascular disorders
Hypertension
Common (>1%, <10%)
Blood and the lymphatic
system disorders
Thromboembolic
event
Common (>1%, <10%)
Nervous system disorders
Headache
Common (>1%, <10%)
-
Patients in an autologous blood predonation programme
Patients in an autologous blood predonation programme have been reported to show a slightly higher
frequency of thromboembolic events. However, a causal relationship with treatment with
NeoRecormon could not be established.
In placebo controlled trials temporary iron deficiency was more pronounced in patients treated with
NeoRecormon than in controls (see section 4.4).
The incidences of undesirable effects in clinical trials, considered related to treatment with
NeoRecoromon are shown in the table below. Within each frequency grouping, undesirable effects are
presented in order of decreasing seriousness.
System Organ Class
Adverse Drug
Reaction
Incidence
Nervous system disorders
Headache
Common (>1%, <10%)
-
All indications
Rarely (≥1/10.000 to ≤1/1.000), epoetin beta treatment-related skin reactions such as rash, pruritus,
urticaria or injection site reactions may occur. In very rare cases (≤1/10.000), epoetin beta treatment-
related anaphylactoid reactions have been reported. However, in controlled clinical studies no
increased incidence of hypersensitivity reactions was found.
In very rare cases (≤1/10.000), particularly when starting treatment, epoetin beta treatment-related flu-
like symptoms such as fever, chills, headaches, pain in the limbs, malaise and/or bone pain have been
reported. These reactions were mild or moderate in nature and subsided after a couple of hours or
days.
Data from a controlled clinical trial with epoetin alfa or darbepoetin alfa, reported an incidence of
stroke as common (≥1/100 to <1/10).
The therapeutic margin of NeoRecormon is very wide. Even at very high serum levels no symptoms of
poisoning have been observed.
35











5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antianaemic, ATC code: B03XA
Epoetin beta is identical in its amino acid and carbohydrate composition to erythropoietin that has
been isolated from the urine of anaemic patients.
Erythropoietin is a glycoprotein that stimulates the formation of erythrocytes from its committed
progenitors. It acts as a mitosis stimulating factor and differentiation hormone.
The biological efficacy of epoetin beta has been demonstrated after intravenous and subcutaneous
administration in various animal models
in vivo
(normal and uraemic rats, polycythaemic mice, dogs).
After administration of epoetin beta, the number of erythrocytes, the Hb values and reticulocyte counts
increase as well as the
59
Fe-incorporation rate.
An increased
3
H-thymidine incorporation in the erythroid nucleated spleen cells has been found
in
vitro
(mouse spleen cell culture) after incubation with epoetin beta.
Investigations in cell cultures of human bone marrow cells showed that epoetin beta stimulates
erythropoiesis specifically and does not affect leucopoiesis. Cytotoxic actions of epoetin beta on bone
marrow or on human skin cells were not detected.
After single dose administration of epoetin beta no effects on behaviour or locomotor activity of mice
and circulatory or respiratory function of dogs were observed.
In a randomised, double-blind, placebo-controlled study of 4,038 CRF patients not on dialysis with
type 2 diabetes and haemoglobin levels ≤ 11 g/dL, patients received either treatment with darbepoetin
alfa to target haemoglobin levels of 13 g/dL or placebo (see section 4.4). The study did not meet either
primary objective of demonstrating a reduction in risk for all-cause mortality, cardiovascular
morbidity, or end stage renal disease (ESRD). Analysis of the individual components of the composite
endpoints showed the following HR (95% CI): death 1.05 (0.92, 1.21), stroke 1.92 (1.38, 2.68),
congestive heart failure (CHF) 0.89 (0.74, 1.08), myocardial infarction (MI) 0.96 (0.75, 1.23),
hospitalisation for myocardial ischaemia 0.84 (0.55, 1.27), ESRD 1.02 (0.87, 1.18).
Erythropoietin is a growth factor that primarily stimulates red cell production. Erythropoietin receptors
may be expressed on the surface of a variety of tumour cells.
Survival and tumour progression have been examined in five large controlled studies involving a total
of 2833 patients, of which four were double-blind placebo-controlled studies and one was an open-
label study. Two of the studies recruited patients who were being treated with chemotherapy. The
target haemoglobin concentration in two studies was >13 g/dl; in the remaining three studies it was 12-
14 g/dl. In the open-label study there was no difference in overall survival between patients treated
with recombinant human erythropoietin and controls. In the four placebo-controlled studies the hazard
ratios for overall survival ranged between 1.25 and 2.47 in favour of controls. These studies have
shown a consistent unexplained statistically significant excess mortality in patients who have anaemia
associated with various common cancers who received recombinant human erythropoietin compared
to controls. Overall survival outcome in the trials could not be satisfactorily explained by differences
in the incidence of thrombosis and related complications between those given recombinant human
erythropoietin and those in the control group.
An individual patient data based meta-analysis, which included data from all 12 controlled clinical
studies in anaemic cancer patients conducted with NeoRecormon (n=2301), showed an overall hazard
ratio point estimate for survival of 1.13 in favour of controls (95 % CI 0.87, 1.46). In patients with
baseline haemoglobin ≤ 10 g/dl (n=899), the hazard ratio point estimate for survival was 0.98 (95 %
CI 0.68 to 1.40). An increased relative risk for thromboembolic events was observed in the overall
population (RR 1.62, 95 % CI: 1.13, 2.31).
36
A patient-level data analysis has also been performed on more than 13,900 cancer patients (chemo-,
radio-, chemoradio- or no therapy) participating in 53 controlled clinical trials involving several
epoetins. Meta-analysis of overall survival data produced a hazard ratio point estimate of 1.06 in
favour of controls (95% CI: 1.00, 1.12; 53 trials and 13,933 patients) and for cancer patients receiving
chemotherapy, the overall survival hazard ratio was 1.04 (95% CI: 0.97, 1.11; 38 trials and 10, 441
patients). Meta-analyses also indicate consistently a significantly increased relative risk of
thromboembolic events in cancer patients receiving recombinant human erythropoietin (see Section
4.4).
In very rare cases, neutralising anti-erythropoietin antibodies with or without pure red cell aplasia
(PRCA) occurred during rHuEPO therapy.
5.2 Pharmacokinetic properties
Pharmacokinetic investigations in healthy volunteers and uraemic patients show that the half-life of
intravenously administered epoetin beta is between 4 and 12 hours and that the distribution volume
corresponds to one to two times the plasma volume. Analogous results have been found in animal
experiments in uraemic and normal rats.
After subcutaneous administration of epoetin beta to uraemic patients, the protracted absorption results
in a serum concentration plateau, whereby the maximum concentration is reached after an average of
12 - 28 hours. The terminal half-life is higher than after intravenous administration, with an average of
13 - 28 hours.
Bioavailability of epoetin beta after subcutaneous administration is between 23 and 42 % as compared
with intravenous administration.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity, genotoxicity, and toxicity to reproduction.
A carcinogenicity study with homologous erythropoietin in mice did not reveal any signs of
proliferative or tumourigenic potential.
6.
6.1 List of excipients
Lyophilisate:
Urea,
Sodium chloride,
Polysorbate 20,
Sodium dihydrogen phosphate,
Disodium hydrogen phosphate,
Calcium chloride,
Glycine,
L-Leucine,
L-Isoleucine,
L-Threonine,
L-Glutamic acid,
L-Phenylalanine.
Solvent:
Benzyl alcohol,
Benzalkonium chloride,
Water for injections.
37
6.2 Incompatibilities
NeoRecormon in cartridge should only be used with the Reco-Pen. In the absence of compatibility
studies, this medicinal product should not be mixed with other medicinal products.
6.3 Shelf life
2 years.
Chemical and physical in-use stability of the reconstituted solution has been demonstrated for one
month at 2°C-8°C. From a microbiological point of view, once opened, the reconstituted solution may
be stored for maximum of one month at 2°C-8°C. Other in-use storage times and conditions are the
responsibility of the user.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C).
Keep the cartridge in the outer carton, in order to protect from light.
For the purpose of ambulatory use, the patient may remove the cartridge not yet inserted into the
Reco-Pen from the refrigerator and store it at room temperatur (not above 25°C) for one single period
of up to 5 days.
After insertion into the Reco-Pen, the cooling chain may only be interrupted for administration of the
product.
For storage conditions of the reconstituted medicinal product see section 6.3.
6.5 Nature and contents of container
Lyophilisate (10,000 IU) and solvent (1 ml) for solution for injection in two-chamber cartridge for
Reco-Pen (type I glass) with front disk made of rubber material of pharmaceutical quality and with
stoppers (teflonised rubber). Pack sizes of 1 or 3.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
This NeoRecormon presentation is a two-chamber cartridge containing lyophilisate for solution for
injection and preserved solution. The ready-to-use solution is prepared by inserting the cartridge into
the Reco-Pen. Prior to this a needle should be attached to the Reco-Pen.
Only solutions which are clear
or slightly opalescent, colourless and practically free of visible particles may be injected.
Please observe the instructions for use which are delivered with the Reco-Pen.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW
United Kingdom
38
8.
EU/1/97/031/021 - 022
9.
Date of the first authorisation: 16 July 1997
Date of the last renewal: 16 July 2007
Detailed information on this medicinal product is available on the website of the European Medicines
Agency http://www.ema.europa.eu/
39
1.
FURTHER INFORMATION
What NeoRecormon contains
-
The active substance is epoetin beta. One pre-filled syringe contains either 500, 1,000, 2,000,
3,000, 4,000, 5,000, 6,000, 10,000, 20,000 or 30,000 IU (international units) epoetin beta in
0.3 ml or 0.6 ml water for injections.
-
The other ingredients are urea, sodium chloride, polysorbate 20, sodium dihydrogen phosphate
dihydrate, disodium phospate dodecahydrate, calcium chloride dihydrate, glycine, L-Leucine,
L-Isoleucine, L-Threonine, L-Glutamic acid, and L-Phenylalanine.
What NeoRecormon looks like and contents of the pack
Colourless, clear to slightly opalescent solution.
NeoRecormon is provided as a solution for injection in 1, 4 or 6 pre-filled syringes with 1, 4 or 6
needles.
Pack size of 1, 4 or 6.
Not all pack sizes may be marketed.
Marketing Authorisation Holder and Manufacturer
Marketing Authorisation Holder
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW
United Kingdom
Manufacturer
Roche Diagnostics GmbH
Sandhofer Str. 116
68305 Mannheim
Germany
For any information about this medicinal product, please contact the local representative of the
Marketing Authorisation Holder:
België/Belgique/Belgien
N.V. Roche S.A.
Tél/Tel: +32 (0) 2 525 82 11
Luxembourg/Luxemburg
(Voir/siehe Belgique/Belgien)
България
Рош България ЕООД
Тел: +359 2 818 44 44
Magyarország
Roche (Magyarország) Kft.
Tel: +36 - 23 446 800
Česká republika
Roche s. r. o.
Tel: +420 - 2 20382111
Malta
(See United Kingdom)
Danmark
Roche a/s
Tlf: +45 - 36 39 99 99
Nederland
Roche Nederland B.V.
Tel: +31 (0) 348 438050
296
Deutschland
Roche Pharma AG
Tel: +49 (0) 7624 140
Norge
Roche Norge AS
Tlf: +47 - 22 78 90 00
Eesti
Roche Eesti OÜ
Tel: + 372 - 6 177 380
Österreich
Roche Austria GmbH
Tel: +43 (0) 1 27739
Ελλάδα
Roche (Hellas) A.E.
Τηλ: +30 210 61 66 100
Polska
Roche Polska Sp.z o.o.
Tel: +48 - 22 345 18 88
España
Roche Farma S.A.
Tel: +34 - 91 324 81 00
Portugal
Roche Farmacêutica Química, Lda
Tel: +351 - 21 425 70 00
France
Roche
Tél: +33 (0) 1 46 40 50 00
România
Roche România S.R.L.
Tel: +40 21 206 47 01
Ireland
Roche Products (Ireland) Ltd.
Tel: +353 (0) 1 469 0700
Slovenija
Roche farmacevtska družba d.o.o.
Tel: +386 - 1 360 26 00
Ísland
Roche a/s
c/o Icepharma hf
Sími: +354 540 8000
Slovenská republika
Roche Slovensko, s.r.o.
Tel: +421 - 2 52638201
Italia
Roche S.p.A.
Tel: +39 - 039 2471
Suomi/Finland
Roche Oy
Puh/Tel: +358 (0) 10 554 500
Kύπρος
Γ.Α.Σταμάτης & Σια Λτδ.
Τηλ: +357 - 22 76 62 76
Sverige
Roche AB
Tel: +46 (0) 8 726 1200
Latvija
Roche Latvija SIA
Tel: +371 - 7 039831
United Kingdom
Roche Products Ltd.
Tel: +44 (0) 1707 366000
Lietuva
UAB “Roche Lietuva”
Tel: +370 5 2546799
This leaflet was last approved in
Detailed information on this medicine is available on the European Medicines Agency web site:
http://www.ema.europa.eu/.
297
Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/neorecormon.html
Copyright © 1995-2021 ITA all rights reserved.