










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Nplate 250 micrograms powder for solution for injection
2.
Each vial contains 250 µg of romiplostim. After reconstitution, a deliverable volume of 0.5 ml
solution contains 250 µg of romiplostim (500 µg/ml). An additional overfill is included in each vial to
ensure that 250 µg of romiplostim can be delivered.
Romiplostim is produced by recombinant DNA technology in
Escherichia coli
(
E. coli
).
For a full list of excipients, see section 6.1.
3.
Powder for solution for injection.
This powder is white.
4.
Nplate is indicated for adult chronic immune (idiopathic) thrombocytopenic purpura (ITP)
splenectomised patients who are refractory to other treatments (e.g. corticosteroids, immunoglobulins).
Nplate may be considered as second line treatment for adult non-splenectomised patients where
surgery is contra-indicated.
Treatment should remain under the supervision of a physician who is experienced in the treatment of
haematological diseases.
Posology
Nplate should be administered once weekly as a subcutaneous injection.
Initial dose
The initial dose of romiplostim is 1 µg/kg based on actual body weight.
Dose calculation
Weight* in kg x Dose in µg/kg = Individual patient dose in µg
Volume to administer: Dose in µg x
1 ml
500 µg
= Amount to inject in ml
2
Initial or subsequent
once weekly dose:





Example:
75 kg patient is initiated at 1 µg/kg of romiplostim.
The individual patient dose =
75 kg x 1 µg = 75 µg
The corresponding amount of Nplate solution to inject =
75 µg x
1 ml
= 0.15 ml
500 µg
*Actual body weight at initiation of treatment should always be used when calculating dose of
romiplostim. Future dose adjustments are based on changes in platelet counts only and made in
1 µg/kg increments (see table below).
Dose adjustments
A subject’s actual body weight at initiation of therapy should be used to calculate dose. The once
weekly dose of romiplostim should be increased by increments of 1 μg/kg until the patient achieves a
platelet count ≥ 50 x 10
9
/l. Platelet counts should be assessed weekly until a stable platelet count
(≥ 50 x 10
9
/l for at least 4 weeks without dose adjustment) has been achieved. Platelet counts should
be assessed monthly thereafter. Do not exceed a maximum once weekly dose of 10 μg/kg.
Adjust the dose as follows:
Platelet count
(x 10
9
/l)
Action
< 50
Increase once weekly dose by 1 μg/kg
consecutive weeks
Decrease once weekly dose by 1 μg/kg
Do not administer, continue to assess the platelet count weekly
> 250
After the platelet count has fallen to < 150 x 10
9
/l, resume dosing with once
weekly dose reduced by 1 μg/kg
Due to the interindividual variable platelet response, in some patients platelet count may abruptly fall
below 50 x 10
9
/l after dose reduction or treatment discontinuation. In these cases, if clinically
appropriate, higher cut-off levels of platelet count for dose reduction (200 x 10
9
/l) and treatment
interruption (400 x 10
9
/l) may be considered according to medical judgement.
A loss of response or failure to maintain a platelet response with romiplostim within the recommended
dosing range should prompt a search for causative factors (see section 4.4, loss of response to
romiplostim).
Treatment discontinuation
Treatment with romiplostim should be discontinued if the platelet count does not increase to a level
sufficient to avoid clinically important bleeding after four weeks of romiplostim therapy at the highest
weekly dose of 10 μg/kg.
Patients should be clinically evaluated periodically and continuation of treatment should be decided on
an individual basis by the treating physician. The reoccurrence of thrombocytopenia is likely upon
discontinuation of treatment (see section 4.4).
Method of administration
For subcutaneous use.
After reconstitution of the powder, Nplate solution for injection is administered subcutaneously. The
injection volume may be very small. A syringe with graduations of 0.01 ml should be used.
3
> 150 for two












For instructions on reconstitution of Nplate before administration, see section 6.6.
Elderly patients (≥ 65 years)
No overall differences in safety or efficacy have been observed in patients < 65 and ≥ 65 years of age
(see section 5.1). Although based on these data no adjustment of the dosing regimen is required for
older patients, care is advised considering the small number of elderly patients included in the clinical
trials so far.
Paediatric population
Nplate is not recommended for use in children below age 18 due to insufficient data on safety or
efficacy. No recommendation on a posology can be made in this population.
Hepatic Impairment
Romiplostim should not be used in patients with moderate to severe hepatic impairment (Child-Pugh
score ≥ 7) unless the expected benefit outweighs the identified risk of portal venous thrombosis in
patients with thrombocytopenia associated to hepatic insufficiency treated with TPO agonists (see
section 4.4).
If the use of romiplostim is deemed necessary, platelet count should be closely monitored to minimise
the risk of thromboembolic complications.
Renal impairment
No formal clinical studies have been conducted in these patient populations. Nplate should be used
with caution in these populations.
Hypersensitivity to the active substance, to any of the excipients or to
E. coli
derived proteins.
The following special warnings and precautions have been actually observed or are potential class
effects based on the pharmacological mechanism of action of thrombopoietin (TPO) receptor
stimulators.
Reoccurrence of thrombocytopenia and bleeding after cessation of treatment
Thrombocytopenia is likely to reoccur upon discontinuation of treatment with romiplostim. There is an
increased risk of bleeding if romiplostim treatment is discontinued in the presence of anticoagulants or
anti-platelet agents. Patients should be closely monitored for a decrease in platelet count and medically
managed to avoid bleeding upon discontinuation of treatment with romiplostim. It is recommended
that, if treatment with romiplostim is discontinued, ITP treatment be restarted according to current
treatment guidelines. Additional medical management may include cessation of anticoagulant and/or
antiplatelet therapy, reversal of anticoagulation, or platelet support.
Increased bone marrow reticulin
Increased bone marrow reticulin is believed to be a result of TPO receptor stimulation, leading to an
increased number of megakaryocytes in the bone marrow, which may subsequently release cytokines.
Increased reticulin may be suggested by morphological changes in the peripheral blood cells and can
be detected through bone marrow biopsy. Therefore, examinations for cellular morphological
abnormalities using peripheral blood smear and complete blood count (CBC) prior to and during
4
treatment with romiplostim are recommended. See section 4.8 for information on the increases of
reticulin observed in romiplostim clinical trials.
If a loss of efficacy and abnormal peripheral blood smear is observed in patients, administration of
romiplostim should be discontinued, a physical examination should be performed, and a bone marrow
biopsy with appropriate staining for reticulin should be considered. If available, comparison to a prior
bone marrow biopsy should be made. If efficacy is maintained and abnormal peripheral blood smear is
observed in patients, the physician should follow appropriate clinical judgment, including
consideration of a bone marrow biopsy, and the risk-benefit of romiplostim and alternative ITP
treatment options should be re-assessed.
Thrombotic/thromboembolic complications
Platelet counts above the normal range present a theoretical risk for thrombotic/thromboembolic
complications. The incidence of thrombotic/thromboembolic events observed in clinical trials was
similar between romiplostim and placebo, and an association between these events and elevated
platelet counts was not observed. Caution should be used when administering romiplostim to patients
with known risk factors for thromboembolism including but not limited to inherited (e.g. Factor V
Leiden) or acquired risk factors (e.g. ATIII deficiency, antiphospholipid syndrome), advanced age,
patients with prolonged periods of immobilisation, malignancies, contraceptives and hormone
replacement therapy, surgery/trauma, obesity and smoking.
Cases of thromboembolic events (TEEs), including portal vein thrombosis, have been reported in
patients with chronic liver disease receiving romiplostim. Romiplostim should be used with caution in
these populations. Dose adjustment guidelines should be followed (see section 4.2).
Progression of existing haematopoietic malignancies or Myelodysplastic Syndromes (MDS)
TPO receptor stimulators are growth factors that lead to thrombopoietic progenitor cell expansion,
differentiation, and platelet production. The TPO receptor is predominantly expressed on the surface
of cells of the myeloid lineage. For TPO receptor stimulators there is a theoretical concern that they
may stimulate the progression of existing haematopoietic malignancies or MDS.
The diagnosis of ITP in adults and elderly patients should have been confirmed by the exclusion of
other clinical entities presenting with thrombocytopenia. Consideration should be given to performing
a bone marrow aspirate and biopsy over the course of the disease and treatment, particularly in
patients over 60 years of age, those with systemic symptoms or abnormal signs.
Romiplostim should not be used for the treatment of thrombocytopenia due to MDS or any other cause
of thrombocytopenia other than ITP outside of clinical trials. The risk-benefit profile for romiplostim
has not been established in MDS or other non-ITP patient populations. In clinical studies of treatment
with romiplostim in patients with MDS, there were reported cases of progression to acute myeloid
leukaemia (AML), however this is an expected clinical outcome of MDS and the relationship to
romiplostim treatment is unclear.
Loss of response to romiplostim
A loss of response or failure to maintain a platelet response with romiplostim treatment within the
recommended dosing range should prompt a search for causative factors, including immunogenicity
(see section 4.8) and increased bone marrow reticulin (see above).
Effects of romiplostim on red and white blood cells
Alterations in red (decrease) and white (increase) blood cell parameters have been observed in non-
clinical toxicology studies (rat and monkey) but not in ITP patients. Monitoring of these parameters
should be considered in patients treated with romiplostim.
5
No interaction studies have been performed. The potential interactions of romiplostim with co-
administered medicinal products due to binding to plasma proteins remain unknown.
Medicinal products used in the treatment of ITP in combination with romiplostim in clinical studies
included corticosteroids, danazol, and/or azathioprine, intravenous immunoglobulin (IVIG), and
anti-D immunoglobulin. Platelet counts should be monitored when combining romiplostim with other
medicinal products for the treatment of ITP in order to avoid platelet counts outside of the
recommended range (see section 4.2).
Corticosteroids, danazol, and azathioprine use may be reduced or discontinued when given in
combination with romiplostim (see section 5.1). Platelet counts should be monitored when reducing or
discontinuing other ITP treatments in order to avoid platelet counts below the recommended range
(see section 4.2).
Pregnancy
For romiplostim no clinical data on exposed pregnancies are available.
Studies in animals have
shown reproductive toxicity, such as transplacental passage and increased
foetal platelet counts in rats (see section 5.3). The potential risk for humans is unknown.
Romiplostim should not be used during pregnancy unless clearly necessary.
Breast-feeding
There are no data on excretion of romiplostim in human milk. However, excretion is likely and a risk
to the suckling child cannot be excluded. A decision on whether to continue/discontinue breast-feeding
or to continue/discontinue therapy with romiplostim should be made taking into account the benefit of
breast-feeding to the child and the benefit of romiplostim therapy to the woman.
No studies on the effects on the ability to drive and use machines have been performed. However,
patients should be informed that in clinical trials mild to moderate, transient bouts of dizziness were
experienced by some patients, which may affect the ability to drive or use machines.
a.
Summary of the safety profile
Based on an analysis of all adult ITP patients receiving romiplostim in 4 controlled and 5 uncontrolled
clinical studies, the overall subject incidence of all adverse reactions for romiplostim-treated subjects
was 91.5% (248/271). The mean duration of exposure to romiplostim in this study population was
50 weeks.
b.
Tabulated list of adverse reactions
Frequencies are defined as: Very common (≥ 1/10), Common (≥ 1/100 to < 1/10) and Uncommon
(≥ 1/1000 to < 1/100). Within each MedDRA system organ class and frequency grouping, undesirable
effects are presented in order of decreasing incidence.
MedDRA system
organ class
Very common Common
Uncommon
6






MedDRA system
organ class
Very common Common
Uncommon
Blood and lymphatic
system disorders
Bone marrow
disorder*
Thrombocytopenia*
Anaemia
Aplastic anaemia
Bone marrow failure
Leukocytosis
Splenomegaly
Thrombocythaemia
Platelet count increased
Platelet count abnormal
Cardiac disorders
Myocardial infarction
Heart rate increased
Ear and labyrinth
disorders
Vertigo
Eye disorders
Conjunctival haemorrhage
Accommodation disorder
Blindness
Eye disorder
Eye pruritus
Lacrimation increased
Papilloedema
Visual disturbances
Gastrointestinal
disorders
Nausea
Diarrhoea
Abdominal pain
Constipation
Dyspepsia
Vomiting
Rectal haemorrhage
Breath odour
Dysphagia
Gastro-oesophageal reflux
disease
Haematochezia
Mouth haemorrhage
Stomach discomfort
Stomatitis
Tooth discolouration
General disorders and
administration site
conditions
Fatigue
Oedema peripheral
Influenza like illness
Pain
Asthenia
Pyrexia
Chills
Injection site reaction
Injection site haemorrhage
Chest pain
Irritability
Malaise
Face oedema
Feeling hot
Feeling jittery
Hepatobiliary
disorders
Portal vein thrombosis
Increase in transaminase
Infections and
infestations
Influenza
Localised infection
Nasopharyngitis
Injury, poisoning and
procedural
complications
Contusion
Investigations
Blood pressure increased
Blood lactate dehydrogenase
increased
Body temperature increased
Weight decreased
Weight increased
7
















MedDRA system
organ class
Very common Common
Uncommon
Metabolism and
nutrition disorders
Alcohol intolerance
Anorexia
Decreased appetite
Dehydration
Gout
Musculoskeletal and
connective tissue
disorders
Arthralgia
Myalgia
Muscle spasms
Pain in extremity
Back pain
Bone pain
Muscle tightness
Muscular weakness
Shoulder pain
Muscle twitching
Neoplasms benign,
malignant and
unspecified (incl cysts
and polyps)
Multiple myeloma
Myelofibrosis
Nervous system
disorders
Headache Dizziness
Migraine
Paraesthesia
Clonus
Dysgeusia
Hypoaesthesia
Hypogeusia
Neuropathy peripheral
Transverse sinus thrombosis
Psychiatric disorders
Insomnia
Depression
Abnormal dreams
Renal and urinary
disorders
Protein urine present
Reproductive system
and breast disorders
Vaginal haemorrhage
Respiratory, thoracic
and mediastinal
disorders
Pulmonary embolism* Cough
Rhinorrhoea
Dry throat
Dyspnoea
Nasal congestion
Painful respiration
Skin and subcutaneous
tissue disorders
Pruritus
Ecchymosis
Rash
Alopecia
Photosensitivity reaction
Acne
Dermatitis contact
Dry skin
Eczema
Erythema
Exfoliative rash
Hair growth abnormal
Prurigo
Purpura
Rash papular
Rash pruritic
Skin nodule
Skin odour abnormal
Urticaria
8















MedDRA system
organ class
Very common Common
Uncommon
Vascular disorders
Flushing
Deep vein thrombosis
Hypotension
Peripheral embolism
Peripheral ischaemia
Phlebitis
Thrombophlebitis superficial
Thrombosis
* see section 4.4
c.
Description of selected adverse reactions
In addition the reactions listed below have been deemed to be related to romiplostim treatment.
Thrombocytosis
Based on an analysis of all adult ITP patients receiving romiplostim in 4 controlled and 5 uncontrolled
clinical studies, 3 events of thrombocytosis were reported, n = 271.
No clinical sequelae were reported
in association with the elevated platelet counts in any of the 3 subjects.
Thrombocytopenia after cessation of treatment
Based on an analysis of all adult ITP patients receiving romiplostim in 4 controlled and 5 uncontrolled
clinical studies, 4 events of thrombocytopenia after cessation of treatment were reported, n = 271 (see
section 4.4).
Increased bone marrow reticulin
In clinical studies, romiplostim treatment was discontinued in 4 of the 271 patients because of bone
marrow reticulin deposition. In 6 additional patients reticulin was observed upon bone marrow biopsy
(see section 4.4).
Immunogenicity
Clinical studies in adult ITP patients examined antibodies to romiplostim.
While 5.8% and 3.9% of the subjects were positive for developing binding antibodies to romiplostim
and TPO respectively, only 2 subjects (0.4%) were positive for neutralizing antibodies to romiplostim
but these antibodies did not cross react with endogenous TPO. Both subjects tested negative for
neutralising antibodies to romiplostim at 4 months after the end of dosing. The incidence of pre-
existing antibodies to romiplostim and TPO was 8.0% and 5.4%, respectively.
As with all therapeutic proteins, there is a potential for immunogenicity. If formation of neutralising
antibodies is suspected, contact the local representative of the Marketing Authorisation Holder (see
section 6 of the Package Leaflet) for antibody testing.
Adverse reactions from spontaneous reporting:
The frequency category of the adverse reactions identified from spontaneous reporting that have not
been reported in clinical trials cannot be estimated (Frequency: not known). The adverse reactions
identified from spontaneous reporting include:
Vascular disorders: Erythromelalgia.
9







No adverse effects were seen in rats given a single dose of 1000 μg/kg or in monkeys after repeated
administration of romiplostim at 500 µg/kg (100 or 50 times the maximum clinical dose of 10 µg/kg,
respectively).
In the event of overdose, platelet counts may increase excessively and result in
thrombotic/thromboembolic complications. If the platelet counts are excessively increased,
discontinue Nplate and monitor platelet counts. Reinitiate treatment with Nplate in accordance with
dosing and administration recommendations (see section 4.2).
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Antihemorrhagics, ATC code: B02BX04
Romiplostim is an Fc-peptide fusion protein (peptibody) that signals and activates intracellular
transcriptional pathways via the thrombopoietin (TPO) receptor (also known as cMpl) to increase
platelet production. The peptibody molecule is comprised of a human immunoglobulin IgG1 Fc
domain, with each single-chain subunit covalently linked at the C-terminus to a peptide chain
containing 2 TPO receptor-binding domains.
Romiplostim has no amino acid sequence homology to endogenous TPO. In pre-clinical and clinical
studies no anti-romiplostim antibodies cross reacted with endogenous TPO.
Clinical data
The safety and efficacy of romiplostim have been evaluated for up to 3 years of continuous treatment.
In clinical studies, treatment with romiplostim resulted in dose-dependent increases in platelet count.
Time to reach the maximum effect on platelet count is approximately 10-14 days, and is independent
of the dose. After a single subcutaneous dose of 1 to 10 µg/kg romiplostim in ITP patients, the peak
platelet count was 1.3 to 14.9 times greater than the baseline platelet count over a 2 to 3 week period
and the response was variable among patients. The platelet counts of ITP patients who received
6 weekly doses of 1 or 3 µg/kg of romiplostim were within the range of 50 to 450 x 10
9
/l for most
patients. Of the 271 patients who received romiplostim in ITP clinical studies, 55 (20%) were age 65
and over, and 27 (10%) were 75 and over. No overall differences in safety or efficacy have been
observed between older and younger patients in the placebo-controlled studies.
Results from pivotal placebo-controlled studies
The safety and efficacy of romiplostim was evaluated in two placebo-controlled, double-blind studies
in adults with ITP who had completed at least one treatment prior to study entry and are representative
of the entire spectrum of such ITP patients.
Study S1 (212) evaluated patients who were non-splenectomised and had an inadequate response or
were intolerant to prior therapies. Patients had been diagnosed with ITP for approximately 2 years at
the time of study entry. Patients had a median of 3 (range, 1 to 7) treatments for ITP prior to study
entry. Prior treatments included corticosteroids (90% of all patients), immunoglobulins (76%),
rituximab (29%), cytotoxic therapies (21%), danazol (11%), and azathioprine (5%). Patients had a
median platelet count of 19 x 10
9
/l at study entry.
Study S2 (105) evaluated patients who were splenectomised and continued to have thrombocytopenia.
Patients had been diagnosed with ITP for approximately 8 years at the time of study entry. In addition
to a splenectomy, patients had a median of 6 (range, 3 to 10) treatments for ITP prior to study entry.
10
Prior treatments included corticosteroids (98% of all patients), immunoglobulins (97%), rituximab
(71%), danazol (37%), cytotoxic therapies (68%), and azathioprine (24%). Patients had a median
platelet count of 14 x 10
9
/l at study entry.
Both studies were similarly designed. Patients (≥ 18 years) were randomised in a 2:1 ratio to receive a
starting dose of romiplostim 1 µg/kg or placebo. Patients received single subcutaneous weekly
injections for 24 weeks. Doses were adjusted to maintain (50 to 200 x 10
9
/l) platelet counts. In both
studies, efficacy was determined by an increase in the proportion of patients who achieved a durable
platelet response. The median average weekly dose for splenectomised patients was 3 µg/kg and for
non-splenectomised patients was 2 µg/kg.
A significantly higher proportion of patients receiving romiplostim achieved a durable platelet
response compared to patients receiving placebo in both studies. Following the first 4-weeks of study
romiplostim maintained platelet counts ≥ 50 x 10
9
/l in between 50% to 70% of patients during the
6 month treatment period in the placebo-controlled studies. In the placebo group, 0% to 7% of patients
were able achieve a platelet count response during the 6 months of treatment. A summary of the key
efficacy endpoints is presented below.
Summary of key efficacy results from placebo-controlled studies
Study 1
non-splenectomised
patients
Study 2
splenectomised patients
Combined
studies 1 & 2
romiplostim
(n = 41)
Placebo
(n = 21)
romiplostim
(n = 42)
Placebo
(n = 21)
romiplostim
(n = 83)
Placebo
(n = 42)
No. (%)
patients
with
durable
platelet
response
a
25 (61%) 1 (5%) 16 (38%) 0 (0%) 41 (50%) 1 (2%)
(95% CI) (45%, 76%) (0%, 24%) (24%, 54%) (0%, 16%) (38%, 61%) (0%, 13%)
p-value
< 0.0001
0.0013
< 0.0001
No. (%)
patients
with
overall
platelet
response
b
36 (88%) 3 (14%) 33 (79%) 0 (0%) 69 (83%) 3 (7%)
(95% CI) (74%, 96%) (3%, 36%) (63%, 90%) (0%, 16%) (73%, 91%) (2%, 20%)
p-value
< 0.0001
< 0.0001
< 0.0001
Mean no.
weeks with
platelet
response
c
15
1
12
0
14
1
(SD)
3.5
7.5
7.9
0.5
7.8
2.5
p-value
< 0.0001
< 0.0001
< 0.0001
No. (%)
patients
requiring
rescue
therapies
d
8(20%) 13 (62%) 11 (26%) 12 (57%) 19 (23%) 25 (60%)
(95% CI) (9%, 35%) (38%, 82%) (14%, 42%) (34%, 78%) (14%, 33%) (43%, 74%)
p-value
0.001
0.0175
< 0.0001
11



















Study 1
non-splenectomised
patients
Study 2
splenectomised patients
Combined
studies 1 & 2
romiplostim
(n = 41)
Placebo
(n = 21)
romiplostim
(n = 42)
Placebo
(n = 21)
romiplostim
(n = 83)
Placebo
(n = 42)
No. (%)
patients
with
durable
platelet
response
with stable
dose
e
21 (51%) 0 (0%) 13 (31%) 0 (0%) 34 (41%) 0 (0%)
(95% CI) (35%, 67%) (0%, 16%) (18%, 47%) (0%, 16%) (30%, 52%) (0%, 8%)
p-value 0.0001 0.0046 < 0.0001
a
Durable platelet response was defined as weekly platelet count ≥ 50 x 10
9
/l for 6 or more times for study
weeks 18-25 in the absence of rescue therapies any time during the treatment period.
b
Overall platelet response is defined as achieving durable or transient platelet responses. Transient platelet
response was defined as weekly platelet count≥ 50 x 10
9
/l for 4 or more times during study weeks 2-25
but wi thout durable p latelet r esponse. P atient may n ot h ave a weekly r esponse wi thin 8 weeks af ter
receiving any rescue medicinal products.
c
Number of weeks with platelet response is defined as number of weeks with platelet counts ≥ 50 x 10
9
/l
during study weeks 2-25. Patient may not have a weekly response within 8 weeks after receiving any
rescue medicinal products.
d
Rescue therapies defined as any therapy administered to raise platelet counts. Patients requiring rescue
medicinal products were not considered for durable platelet response. Rescue therapies allowed in the
study were IVIG, platelet transfusions, anti-D immunoglobulin, and corticosteroids.
e
Stable dose defined as dose maintained within ± 1 µg/kg during the last 8 weeks of treatment.
Reduction in permitted concurrent ITP medical therapies
In both placebo-controlled, double-blind studies, patients already receiving ITP medical therapies at a
constant dosing schedule were allowed to continue receiving these medical treatments throughout the
study (corticosteroids, danazol and/or azathioprine). Twenty-one non-splenectomised and
18 splenectomised patients received on-study ITP medical treatments (primarily corticosteroids) at the
start of study. All (100%) splenectomised patients who were receiving romiplostim were able to
reduce the dose by more than 25% or discontinue the concurrent ITP medical therapies by the end of
the treatment period compared to 17% of placebo treated patients. Seventy-three percent of
non-splenectomised patients receiving romiplostim were able to reduce the dose by more than 25% or
discontinue concurrent ITP medical therapies by the end of the study compared to 50% of placebo
treated patients (see section 4.5).
Bleeding events
Across the entire ITP clinical programme an inverse relationship between bleeding events and platelet
counts was observed. All clinically significant (≥ grade 3) bleeding events occurred at platelet counts
< 30 x 10
9
/l. All bleeding events ≥ grade 2 occurred at platelet counts < 50 x 10
9
/l. No statistically
significant differences in the overall incidence of bleeding events were observed between Nplate and
placebo treated patients.
In the two placebo-controlled studies, 9 patients reported a bleeding event that was considered serious
(5 [6.0%] romiplostim, 4 [9.8%] placebo; Odds Ratio [romiplostim/placebo] = 0.59; 95% CI = (0.15,
2.31)). Bleeding events that were grade 2 or higher were reported by 15% of patients treated with
romiplostim and 34% of patients treated with placebo (Odds Ratio; [romiplostim/placebo] = 0.35; 95%
CI = (0.14, 0.85)).
12














5.2 Pharmacokinetic properties
The pharmacokinetics of romiplostim involved target-mediated disposition, which is presumably
mediated by TPO receptors on platelets and other cells of the thrombopoietic lineage such as
megakaryocytes.
Absorption
After subcutaneous administration of 3 to 15 μg/kg romiplostim, maximum romiplostim serum levels
in ITP patients were obtained after 7-50 hours (median 14 hours). The serum concentrations varied
among patients and did not correlate with the dose administered. Romiplostim serum levels appear
inversely related to platelet counts.
Distribution
The volume of distribution of romiplostim following intravenous administration of romiplostim
decreased nonlinearly from 122, 78.8, to 48.2 ml/kg for intravenous doses of 0.3, 1.0 and 10 μg/kg,
respectively in healthy subjects. This non-linear decrease in volume of distribution is in line with the
(megakaryocyte and platelet) target-mediated binding of romiplostim, which may be saturated at the
higher doses applied.
Elimination
Elimination half-life of romiplostim in ITP patients ranged from 1 to 34 days (median, 3.5 days).
The elimination of serum romiplostim is in part dependent on the TPO receptor on platelets. As a
result for a given dose, patients with high platelet counts are associated with low serum concentrations
and
vice versa
. In another ITP clinical study, no accumulation in serum concentrations was observed
after 6 weekly doses of romiplostim (3 μg/kg).
Special patient populations
Pharmacokinetics of romiplostim in patients with renal and hepatic impairment has not been
investigated. Romiplostim pharmacokinetics appear not affected by age, weight and gender to a
clinically significant extent.
5.3 Preclinical safety data
Multiple dose romiplostim toxicology studies were conducted in rats for 4 weeks and in monkeys for
up to 6 months. In general, effects observed during these studies were related to the thrombopoietic
activity of romiplostim and were similar regardless of study duration. Injection site reactions were also
related to romiplostim administration. Myelofibrosis has been observed in the bone marrow of rats at
all tested dose levels. In these studies, myelofibrosis was not observed in animals after a 4-week post-
treatment recovery period, indicating reversibility.
In 1-month rat and monkey toxicology studies, a mild decrease in red blood cell count, haematocrit
and haemoglobin was observed. There was also a stimulatory effect on leukocyte production, as
peripheral blood counts for neutrophils, lymphocytes, monocytes, and eosinophils were mildly
increased. In the longer duration chronic monkey study, there was no effect on the erythroid and
leukocytic lineages when romiplostim was administered for 6 months where the administration of
romiplostim was decreased from thrice weekly to once weekly. Additionally, in the phase 3 pivotal
studies, romiplostim did not affect the red blood cell and white blood cells lineages relative to placebo
treated subjects.
Due to the formation of neutralising antibodies pharmacodynamic effects of romiplostim in rats were
often decreasing at prolonged duration of administration. Toxicokinetic studies showed no interaction
of the antibodies with the measured concentrations. Although high doses were tested in the animal
studies, due to differences between the laboratory species and humans with regard to the sensitivity for
13
the pharmacodynamic effect of romiplostim and the effect of neutralising antibodies, safety margins
cannot be reliably estimated.
Carcinogenesis:
The carcinogenic potential of romiplostim has not been evaluated. Therefore, the risk
of potential carcinogenicity of romiplostim in humans remains unknown.
Reproductive toxicology:
In all developmental studies neutralising antibodies were formed, which may
have inhibited romiplostim effects. In embryo-foetal development studies in mice and rats, reductions
in maternal body weight were found only in mice. In mice there was evidence of increased post-
implantation loss. In a prenatal and postnatal development study in rats an increase of the duration of
gestation and a slight increase in the incidence of peri-natal pup mortality was found. Romiplostim is
known to cross the placental barrier in rats and may be transmitted from the mother to the developing
foetus and stimulate foetal platelet production. Romiplostim had no observed effect on the fertility of
rats.
6.
6.1 List of excipients
Mannitol (E421)
Sucrose
L-histidine
Hydrochloric acid (for pH adjustment)
Polysorbate 20
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products, except those mentioned in section 6.6.
6.3 Shelf life
3 years.
After reconstitution: Chemical and physical in-use stability has been demonstrated for 24 hours at
25°C and for 24 hours at 2°C – 8°C, when protected from light and kept in the original vial.
From a microbiological point of view, the product should be used immediately. If not used
immediately, in-use storage times and conditions prior to use are the responsibility of the user and
would normally not be longer than 24 hours at 25°C or 24 hours in a refrigerator (2°C – 8°C),
protected from light.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C).
Do not freeze.
Store in the original carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
6.5 Nature and contents of container
5 ml vial (type 1 clear glass) with a stopper (chlorobutyl rubber), seal (aluminium) and a flip-off cap
(polypropylene).
Carton containing 1 or 4 vials of 250 µg of romiplostim.
14
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
Nplate is a sterile but unpreserved product and is intended for single use only. Nplate should be
reconstituted in accordance with good aseptic practice.
Nplate 250 micrograms powder for solution for injection should be reconstituted with 0.72 ml sterile
water for injections, yielding a deliverable volume of 0.5 ml. An additional overfill is included in each
vial to ensure that 250 µg of romiplostim can be delivered.
Sodium chloride solutions or bacteriostatic water should not be used when reconstituting the medicinal
product.
Water for injections should be injected into the vial. The vial contents may be swirled gently and
inverted during dissolution.
The vial should not be shaken or vigorously agitated.
Generally,
dissolution of Nplate takes less than 2 minutes. Visually inspect the solution for particulate matter and
discolouration before administration. The reconstituted solution should be clear and colourless and
should not be administered if particulate matter and/or discolouration are observed.
For the storage condition of the reconstituted product see section 6.3.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
The Netherlands
8.
EU/1/08/497/001
EU/1/08/497/003
9.
4 February 2009
Detailed information on this product is available on the website of the European Medicines Agency
15
1.
Nplate 500 micrograms powder for solution for injection
2.
Each vial contains 500 µg of romiplostim. After reconstitution, a deliverable volume of 1 ml solution
contains 500 µg of romiplostim (500 µg/ml). An additional overfill is included in each vial to ensure
that 500 µg of romiplostim can be delivered.
Romiplostim is produced by recombinant DNA technology in
Escherichia coli
(
E. coli
).
For a full list of excipients, see section 6.1.
3.
Powder for solution for injection.
This powder is white.
4.
Nplate is indicated for adult chronic immune (idiopathic) thrombocytopenic purpura (ITP)
splenectomised patients who are refractory to other treatments (e.g. corticosteroids, immunoglobulins).
Nplate may be considered as second line treatment for adult non-splenectomised patients where
surgery is contra-indicated.
Treatment should remain under the supervision of a physician who is experienced in the treatment of
haematological diseases.
Posology
Nplate should be administered once weekly as a subcutaneous injection.
Initial dose
The initial dose of romiplostim is 1 µg/kg based on actual body weight.
Dose calculation
Weight* in kg x Dose in µg/kg = Individual patient dose in µg
Volume to administer: Dose in µg x
1 ml
500 µg
= Amount to inject in ml
16
Initial or subsequent
once weekly dose:





Example:
75 kg patient is initiated at 1 µg/kg of romiplostim.
The individual patient dose =
75 kg x 1 µg = 75 µg
The corresponding amount of Nplate solution to inject =
75 µg x
1 ml
= 0.15 ml
500 µg
*Actual body weight at initiation of treatment should always be used when calculating dose of
romiplostim. Future dose adjustments are based on changes in platelet counts only and made in
1 µg/kg increments (see table below).
Dose adjustments
A subject’s actual body weight at initiation of therapy should be used to calculate dose. The once
weekly dose of romiplostim should be increased by increments of 1 μg/kg until the patient achieves a
platelet count ≥ 50 x 10
9
/l. Platelet counts should be assessed weekly until a stable platelet count
(≥ 50 x 10
9
/l for at least 4 weeks without dose adjustment) has been achieved. Platelet counts should
be assessed monthly thereafter. Do not exceed a maximum once weekly dose of 10 μg/kg.
Adjust the dose as follows:
Platelet count
(x 10
9
/l)
Action
< 50
Increase once weekly dose by 1 μg/kg
consecutive weeks
Decrease once weekly dose by 1 μg/kg
Do not administer, continue to assess the platelet count weekly
> 250
After the platelet count has fallen to < 150 x 10
9
/l, resume dosing with once
weekly dose reduced by 1 μg/kg
Due to the interindividual variable platelet response, in some patients platelet count may abruptly fall
below 50 x 10
9
/l after dose reduction or treatment discontinuation. In these cases, if clinically
appropriate, higher cut-off levels of platelet count for dose reduction (200 x 10
9
/l) and treatment
interruption (400 x 10
9
/l) may be considered according to medical judgement.
A loss of response or failure to maintain a platelet response with romiplostim within the recommended
dosing range should prompt a search for causative factors (see section 4.4, loss of response to
romiplostim).
Treatment discontinuation
Treatment with romiplostim should be discontinued if the platelet count does not increase to a level
sufficient to avoid clinically important bleeding after four weeks of romiplostim therapy at the highest
weekly dose of 10 μg/kg.
Patients should be clinically evaluated periodically and continuation of treatment should be decided on
an individual basis by the treating physician. The reoccurrence of thrombocytopenia is likely upon
discontinuation of treatment (see section 4.4).
Method of administration
For subcutaneous use.
After reconstitution of the powder, Nplate solution for injection is administered subcutaneously. The
injection volume may be very small. A syringe with graduations of 0.01 ml should be used.
17
> 150 for two












For instructions on reconstitution of Nplate before administration, see section 6.6.
Elderly patients (≥ 65 years)
No overall differences in safety or efficacy have been observed in patients < 65 and ≥ 65 years of age
(see section 5.1). Although based on these data no adjustment of the dosing regimen is required for
older patients, care is advised considering the small number of elderly patients included in the clinical
trials so far.
Paediatric population
Nplate is not recommended for use in children below age 18 due to insufficient data on safety or
efficacy. No recommendation on a posology can be made in this population.
Hepatic Impairment
Romiplostim should not be used in patients with moderate to severe hepatic impairment (Child-Pugh
score ≥ 7) unless the expected benefit outweighs the identified risk of portal venous thrombosis in
patients with thrombocytopenia associated to hepatic insufficiency treated with TPO agonists (see
section 4.4).
If the use of romiplostim is deemed necessary, platelet count should be closely monitored to minimise
the risk of thromboembolic complications.
Renal impairment
No formal clinical studies have been conducted in these patient populations. Nplate should be used
with caution in these populations.
Hypersensitivity to the active substance, to any of the excipients or to
E. coli
derived proteins.
The following special warnings and precautions have been actually observed or are potential class
effects based on the pharmacological mechanism of action of thrombopoietin (TPO) receptor
stimulators.
Reoccurrence of thrombocytopenia and bleeding after cessation of treatment
Thrombocytopenia is likely to reoccur upon discontinuation of treatment with romiplostim. There is an
increased risk of bleeding if romiplostim treatment is discontinued in the presence of anticoagulants or
anti-platelet agents. Patients should be closely monitored for a decrease in platelet count and medically
managed to avoid bleeding upon discontinuation of treatment with romiplostim. It is recommended
that, if treatment with romiplostim is discontinued, ITP treatment be restarted according to current
treatment guidelines. Additional medical management may include cessation of anticoagulant and/or
antiplatelet therapy, reversal of anticoagulation, or platelet support.
Increased bone marrow reticulin
Increased bone marrow reticulin is believed to be a result of TPO receptor stimulation, leading to an
increased number of megakaryocytes in the bone marrow, which may subsequently release cytokines.
Increased reticulin may be suggested by morphological changes in the peripheral blood cells and can
be detected through bone marrow biopsy. Therefore, examinations for cellular morphological
abnormalities using peripheral blood smear and complete blood count (CBC) prior to and during
18
treatment with romiplostim are recommended. See section 4.8 for information on the increases of
reticulin observed in romiplostim clinical trials.
If a loss of efficacy and abnormal peripheral blood smear is observed in patients, administration of
romiplostim should be discontinued, a physical examination should be performed, and a bone marrow
biopsy with appropriate staining for reticulin should be considered. If available, comparison to a prior
bone marrow biopsy should be made. If efficacy is maintained and abnormal peripheral blood smear is
observed in patients, the physician should follow appropriate clinical judgment, including
consideration of a bone marrow biopsy, and the risk-benefit of romiplostim and alternative ITP
treatment options should be re-assessed.
Thrombotic/thromboembolic complications
Platelet counts above the normal range present a theoretical risk for thrombotic/thromboembolic
complications. The incidence of thrombotic/thromboembolic events observed in clinical trials was
similar between romiplostim and placebo, and an association between these events and elevated
platelet counts was not observed. Caution should be used when administering romiplostim to patients
with known risk factors for thromboembolism including but not limited to inherited (e.g. Factor V
Leiden) or acquired risk factors (e.g. ATIII deficiency, antiphospholipid syndrome), advanced age,
patients with prolonged periods of immobilisation, malignancies, contraceptives and hormone
replacement therapy, surgery/trauma, obesity and smoking.
Cases of thromboembolic events (TEEs), including portal vein thrombosis, have been reported in
patients with chronic liver disease receiving romiplostim. Romiplostim should be used with caution in
these populations. Dose adjustment guidelines should be followed (see section 4.2).
Progression of existing haematopoietic malignancies or Myelodysplastic Syndromes (MDS)
TPO receptor stimulators are growth factors that lead to thrombopoietic progenitor cell expansion,
differentiation, and platelet production. The TPO receptor is predominantly expressed on the surface
of cells of the myeloid lineage. For TPO receptor stimulators there is a theoretical concern that they
may stimulate the progression of existing haematopoietic malignancies or MDS.
The diagnosis of ITP in adults and elderly patients should have been confirmed by the exclusion of
other clinical entities presenting with thrombocytopenia. Consideration should be given to performing
a bone marrow aspirate and biopsy over the course of the disease and treatment, particularly in
patients over 60 years of age, those with systemic symptoms or abnormal signs.
Romiplostim should not be used for the treatment of thrombocytopenia due to MDS or any other cause
of thrombocytopenia other than ITP outside of clinical trials. The risk-benefit profile for romiplostim
has not been established in MDS or other non-ITP patient populations. In clinical studies of treatment
with romiplostim in patients with MDS, there were reported cases of progression to acute myeloid
leukaemia (AML), however this is an expected clinical outcome of MDS and the relationship to
romiplostim treatment is unclear.
Loss of response to romiplostim
A loss of response or failure to maintain a platelet response with romiplostim treatment within the
recommended dosing range should prompt a search for causative factors, including immunogenicity
(see section 4.8) and increased bone marrow reticulin (see above).
Effects of romiplostim on red and white blood cells
Alterations in red (decrease) and white (increase) blood cell parameters have been observed in non-
clinical toxicology studies (rat and monkey) but not in ITP patients. Monitoring of these parameters
should be considered in patients treated with romiplostim.
19
No interaction studies have been performed. The potential interactions of romiplostim with co-
administered medicinal products due to binding to plasma proteins remain unknown.
Medicinal products used in the treatment of ITP in combination with romiplostim in clinical studies
included corticosteroids, danazol, and/or azathioprine, intravenous immunoglobulin (IVIG), and
anti-D immunoglobulin. Platelet counts should be monitored when combining romiplostim with other
medicinal products for the treatment of ITP in order to avoid platelet counts outside of the
recommended range (see section 4.2).
Corticosteroids, danazol, and azathioprine use may be reduced or discontinued when given in
combination with romiplostim (see section 5.1). Platelet counts should be monitored when reducing or
discontinuing other ITP treatments in order to avoid platelet counts below the recommended range
(see section 4.2).
Pregnancy
For romiplostim no clinical data on exposed pregnancies are available.
Studies in animals have
shown reproductive toxicity, such as transplacental passage and increased
foetal platelet counts in rats (see section 5.3). The potential risk for humans is unknown.
Romiplostim should not be used during pregnancy unless clearly necessary.
Breast-feeding
There are no data on excretion of romiplostim in human milk. However, excretion is likely and a risk
to the suckling child cannot be excluded. A decision on whether to continue/discontinue breast-feeding
or to continue/discontinue therapy with romiplostim should be made taking into account the benefit of
breast-feeding to the child and the benefit of romiplostim therapy to the woman.
No studies on the effects on the ability to drive and use machines have been performed. However,
patients should be informed that in clinical trials mild to moderate, transient bouts of dizziness were
experienced by some patients, which may affect the ability to drive or use machines.
a.
Summary of the safety profile
Based on an analysis of all adult ITP patients receiving romiplostim in 4 controlled and 5 uncontrolled
clinical studies, the overall subject incidence of all adverse reactions for romiplostim-treated subjects
was 91.5% (248/271). The mean duration of exposure to romiplostim in this study population was
50 weeks.
b.
Tabulated list of adverse reactions
Frequencies are defined as: Very common (≥ 1/10), Common (≥ 1/100 to < 1/10) and Uncommon
(≥ 1/1000 to < 1/100). Within each MedDRA system organ class and frequency grouping, undesirable
effects are presented in order of decreasing incidence.
MedDRA system
organ class
Very common Common
Uncommon
20






MedDRA system
organ class
Very common Common
Uncommon
Blood and lymphatic
system disorders
Bone marrow
disorder*
Thrombocytopenia*
Anaemia
Aplastic anaemia
Bone marrow failure
Leukocytosis
Splenomegaly
Thrombocythaemia
Platelet count increased
Platelet count abnormal
Cardiac disorders
Myocardial infarction
Heart rate increased
Ear and labyrinth
disorders
Vertigo
Eye disorders
Conjunctival haemorrhage
Accommodation disorder
Blindness
Eye disorder
Eye pruritus
Lacrimation increased
Papilloedema
Visual disturbances
Gastrointestinal
disorders
Nausea
Diarrhoea
Abdominal pain
Constipation
Dyspepsia
Vomiting
Rectal haemorrhage
Breath odour
Dysphagia
Gastro-oesophageal reflux
disease
Haematochezia
Mouth haemorrhage
Stomach discomfort
Stomatitis
Tooth discolouration
General disorders and
administration site
conditions
Fatigue
Oedema peripheral
Influenza like illness
Pain
Asthenia
Pyrexia
Chills
Injection site reaction
Injection site haemorrhage
Chest pain
Irritability
Malaise
Face oedema
Feeling hot
Feeling jittery
Hepatobiliary
disorders
Portal vein thrombosis
Increase in transaminase
Infections and
infestations
Influenza
Localised infection
Nasopharyngitis
Injury, poisoning and
procedural
complications
Contusion
Investigations
Blood pressure increased
Blood lactate dehydrogenase
increased
Body temperature increased
Weight decreased
Weight increased
21
















MedDRA system
organ class
Very common Common
Uncommon
Metabolism and
nutrition disorders
Alcohol intolerance
Anorexia
Decreased appetite
Dehydration
Gout
Musculoskeletal and
connective tissue
disorders
Arthralgia
Myalgia
Muscle spasms
Pain in extremity
Back pain
Bone pain
Muscle tightness
Muscular weakness
Shoulder pain
Muscle twitching
Neoplasms benign,
malignant and
unspecified (incl cysts
and polyps)
Multiple myeloma
Myelofibrosis
Nervous system
disorders
Headache Dizziness
Migraine
Paraesthesia
Clonus
Dysgeusia
Hypoaesthesia
Hypogeusia
Neuropathy peripheral
Transverse sinus thrombosis
Psychiatric disorders
Insomnia
Depression
Abnormal dreams
Renal and urinary
disorders
Protein urine present
Reproductive system
and breast disorders
Vaginal haemorrhage
Respiratory, thoracic
and mediastinal
disorders
Pulmonary embolism* Cough
Rhinorrhoea
Dry throat
Dyspnoea
Nasal congestion
Painful respiration
Skin and subcutaneous
tissue disorders
Pruritus
Ecchymosis
Rash
Alopecia
Photosensitivity reaction
Acne
Dermatitis contact
Dry skin
Eczema
Erythema
Exfoliative rash
Hair growth abnormal
Prurigo
Purpura
Rash papular
Rash pruritic
Skin nodule
Skin odour abnormal
Urticaria
22















MedDRA system
organ class
Very common Common
Uncommon
Vascular disorders
Flushing
Deep vein thrombosis
Hypotension
Peripheral embolism
Peripheral ischaemia
Phlebitis
Thrombophlebitis superficial
Thrombosis
* see section 4.4
c.
Description of selected adverse reactions
In addition the reactions listed below have been deemed to be related to romiplostim treatment.
Thrombocytosis
Based on an analysis of all adult ITP patients receiving romiplostim in 4 controlled and 5 uncontrolled
clinical studies, 3 events of thrombocytosis were reported, n = 271.
No clinical sequelae were reported
in association with the elevated platelet counts in any of the 3 subjects.
Thrombocytopenia after cessation of treatment
Based on an analysis of all adult ITP patients receiving romiplostim in 4 controlled and 5 uncontrolled
clinical studies, 4 events of thrombocytopenia after cessation of treatment were reported, n = 271 (see
section 4.4).
Increased bone marrow reticulin
In clinical studies, romiplostim treatment was discontinued in 4 of the 271 patients because of bone
marrow reticulin deposition. In 6 additional patients reticulin was observed upon bone marrow biopsy
(see section 4.4).
Immunogenicity
Clinical studies in adult ITP patients examined antibodies to romiplostim.
While 5.8% and 3.9% of the subjects were positive for developing binding antibodies to romiplostim
and TPO respectively, only 2 subjects (0.4%) were positive for neutralizing antibodies to romiplostim
but these antibodies did not cross react with endogenous TPO. Both subjects tested negative for
neutralising antibodies to romiplostim at 4 months after the end of dosing. The incidence of pre-
existing antibodies to romiplostim and TPO was 8.0% and 5.4%, respectively.
As with all therapeutic proteins, there is a potential for immunogenicity. If formation of neutralising
antibodies is suspected, contact the local representative of the Marketing Authorisation Holder (see
section 6 of the Package Leaflet) for antibody testing.
Adverse reactions from spontaneous reporting:
The frequency category of the adverse reactions identified from spontaneous reporting that have not
been reported in clinical trials cannot be estimated (Frequency: not known). The adverse reactions
identified from spontaneous reporting include:
Vascular disorders: Erythromelalgia.
23







No adverse effects were seen in rats given a single dose of 1000 μg/kg or in monkeys after repeated
administration of romiplostim at 500 µg/kg (100 or 50 times the maximum clinical dose of 10 µg/kg,
respectively).
In the event of overdose, platelet counts may increase excessively and result in
thrombotic/thromboembolic complications. If the platelet counts are excessively increased,
discontinue Nplate and monitor platelet counts. Reinitiate treatment with Nplate in accordance with
dosing and administration recommendations (see section 4.2).
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Antihemorrhagics, ATC code: B02BX04
Romiplostim is an Fc-peptide fusion protein (peptibody) that signals and activates intracellular
transcriptional pathways via the thrombopoietin (TPO) receptor (also known as cMpl) to increase
platelet production. The peptibody molecule is comprised of a human immunoglobulin IgG1 Fc
domain, with each single-chain subunit covalently linked at the C-terminus to a peptide chain
containing 2 TPO receptor-binding domains.
Romiplostim has no amino acid sequence homology to endogenous TPO. In pre-clinical and clinical
studies no anti-romiplostim antibodies cross reacted with endogenous TPO.
Clinical data
The safety and efficacy of romiplostim have been evaluated for up to 3 years of continuous treatment.
In clinical studies, treatment with romiplostim resulted in dose-dependent increases in platelet count.
Time to reach the maximum effect on platelet count is approximately 10-14 days, and is independent
of the dose. After a single subcutaneous dose of 1 to 10 µg/kg romiplostim in ITP patients, the peak
platelet count was 1.3 to 14.9 times greater than the baseline platelet count over a 2 to 3 week period
and the response was variable among patients. The platelet counts of ITP patients who received
6 weekly doses of 1 or 3 µg/kg of romiplostim were within the range of 50 to 450 x 10
9
/l for most
patients. Of the 271 patients who received romiplostim in ITP clinical studies, 55 (20%) were age 65
and over, and 27 (10%) were 75 and over. No overall differences in safety or efficacy have been
observed between older and younger patients in the placebo-controlled studies.
Results from pivotal placebo-controlled studies
The safety and efficacy of romiplostim was evaluated in two placebo-controlled, double-blind studies
in adults with ITP who had completed at least one treatment prior to study entry and are representative
of the entire spectrum of such ITP patients.
Study S1 (212) evaluated patients who were non-splenectomised and had an inadequate response or
were intolerant to prior therapies. Patients had been diagnosed with ITP for approximately 2 years at
the time of study entry. Patients had a median of 3 (range, 1 to 7) treatments for ITP prior to study
entry. Prior treatments included corticosteroids (90% of all patients), immunoglobulins (76%),
rituximab (29%), cytotoxic therapies (21%), danazol (11%), and azathioprine (5%). Patients had a
median platelet count of 19 x 10
9
/l at study entry.
Study S2 (105) evaluated patients who were splenectomised and continued to have thrombocytopenia.
Patients had been diagnosed with ITP for approximately 8 years at the time of study entry. In addition
to a splenectomy, patients had a median of 6 (range, 3 to 10) treatments for ITP prior to study entry.
24
Prior treatments included corticosteroids (98% of all patients), immunoglobulins (97%), rituximab
(71%), danazol (37%), cytotoxic therapies (68%), and azathioprine (24%). Patients had a median
platelet count of 14 x 10
9
/l at study entry.
Both studies were similarly designed. Patients (≥ 18 years) were randomised in a 2:1 ratio to receive a
starting dose of romiplostim 1 µg/kg or placebo. Patients received single subcutaneous weekly
injections for 24 weeks. Doses were adjusted to maintain (50 to 200 x 10
9
/l) platelet counts. In both
studies, efficacy was determined by an increase in the proportion of patients who achieved a durable
platelet response. The median average weekly dose for splenectomised patients was 3 µg/kg and for
non-splenectomised patients was 2 µg/kg.
A significantly higher proportion of patients receiving romiplostim achieved a durable platelet
response compared to patients receiving placebo in both studies. Following the first 4-weeks of study
romiplostim maintained platelet counts ≥ 50 x 10
9
/l in between 50% to 70% of patients during the
6 month treatment period in the placebo-controlled studies. In the placebo group, 0% to 7% of patients
were able achieve a platelet count response during the 6 months of treatment. A summary of the key
efficacy endpoints is presented below.
Summary of key efficacy results from placebo-controlled studies
Study 1
non-splenectomised
patients
Study 2
splenectomised patients
Combined
studies 1 & 2
romiplostim
(n = 41)
Placebo
(n = 21)
romiplostim
(n = 42)
Placebo
(n = 21)
romiplostim
(n = 83)
Placebo
(n = 42)
No. (%)
patients
with
durable
platelet
response
a
25 (61%) 1 (5%) 16 (38%) 0 (0%) 41 (50%) 1 (2%)
(95% CI) (45%, 76%) (0%, 24%) (24%, 54%) (0%, 16%) (38%, 61%) (0%, 13%)
p-value
< 0.0001
0.0013
< 0.0001
No. (%)
patients
with
overall
platelet
response
b
36 (88%) 3 (14%) 33 (79%) 0 (0%) 69 (83%) 3 (7%)
(95% CI) (74%, 96%) (3%, 36%) (63%, 90%) (0%, 16%) (73%, 91%) (2%, 20%)
p-value
< 0.0001
< 0.0001
< 0.0001
Mean no.
weeks with
platelet
response
c
15
1
12
0
14
1
(SD)
3.5
7.5
7.9
0.5
7.8
2.5
p-value
< 0.0001
< 0.0001
< 0.0001
No. (%)
patients
requiring
rescue
therapies
d
8(20%) 13 (62%) 11 (26%) 12 (57%) 19 (23%) 25 (60%)
(95% CI) (9%, 35%) (38%, 82%) (14%, 42%) (34%, 78%) (14%, 33%) (43%, 74%)
p-value
0.001
0.0175
< 0.0001
25



















Study 1
non-splenectomised
patients
Study 2
splenectomised patients
Combined
studies 1 & 2
romiplostim
(n = 41)
Placebo
(n = 21)
romiplostim
(n = 42)
Placebo
(n = 21)
romiplostim
(n = 83)
Placebo
(n = 42)
No. (%)
patients
with
durable
platelet
response
with stable
dose
e
21 (51%) 0 (0%) 13 (31%) 0 (0%) 34 (41%) 0 (0%)
(95% CI) (35%, 67%) (0%, 16%) (18%, 47%) (0%, 16%) (30%, 52%) (0%, 8%)
p-value 0.0001 0.0046 < 0.0001
a
Durable platelet response was defined as weekly platelet count ≥ 50 x 10
9
/l for 6 or more times for study
weeks 18-25 in the absence of rescue therapies any time during the treatment period.
b
Overall platelet response is defined as achieving durable or transient platelet responses. Transient platelet
response was defined as weekly platelet count ≥ 50 x 10
9
/l for 4 or more times during study weeks 2-25
but without durable platelet response. Patient may not have a weekly response within 8 weeks after
receiving any rescue medicinal products.
c
Number of weeks with platelet response is defined as number of weeks with platelet counts ≥ 50 x 10
9
/l
during study weeks 2-25. Patient may not have a weekly response within 8 weeks after receiving any
rescue medicinal products.
d
Rescue therapies defined as any therapy administered to raise platelet counts. Patients requiring rescue
medicinal products were not considered for durable platelet response. Rescue therapies allowed in the
study were IVIG, platelet transfusions, anti-D immunoglobulin, and corticosteroids.
e
Stable dose defined as dose maintained within ± 1 µg/kg during the last 8 weeks of treatment.
Reduction in permitted concurrent ITP medical therapies
In both placebo-controlled, double-blind studies, patients already receiving ITP medical therapies at a
constant dosing schedule were allowed to continue receiving these medical treatments throughout the
study (corticosteroids, danazol and/or azathioprine). Twenty-one non-splenectomised and
18 splenectomised patients received on-study ITP medical treatments (primarily corticosteroids) at the
start of study. All (100%) splenectomised patients who were receiving romiplostim were able to
reduce the dose by more than 25% or discontinue the concurrent ITP medical therapies by the end of
the treatment period compared to 17% of placebo treated patients. Seventy-three percent of
non-splenectomised patients receiving romiplostim were able to reduce the dose by more than 25% or
discontinue concurrent ITP medical therapies by the end of the study compared to 50% of placebo
treated patients (see section 4.5).
Bleeding events
Across the entire ITP clinical programme an inverse relationship between bleeding events and platelet
counts was observed. All clinically significant (≥ grade 3) bleeding events occurred at platelet counts
< 30 x 10
9
/l. All bleeding events ≥ grade 2 occurred at platelet counts < 50 x 10
9
/l. No statistically
significant differences in the overall incidence of bleeding events were observed between Nplate and
placebo treated patients.
In the two placebo-controlled studies, 9 patients reported a bleeding event that was considered serious
(5 [6.0%] romiplostim, 4 [9.8%] placebo; Odds Ratio [romiplostim/placebo] = 0.59; 95% CI = (0.15,
2.31)). Bleeding events that were grade 2 or higher were reported by 15% of patients treated with
romiplostim and 34% of patients treated with placebo (Odds Ratio; [romiplostim/placebo] = 0.35; 95%
CI = (0.14, 0.85)).
26













5.2 Pharmacokinetic properties
The pharmacokinetics of romiplostim involved target-mediated disposition, which is presumably
mediated by TPO receptors on platelets and other cells of the thrombopoietic lineage such as
megakaryocytes.
Absorption
After subcutaneous administration of 3 to 15 μg/kg romiplostim, maximum romiplostim serum levels
in ITP patients were obtained after 7-50 hours (median 14 hours). The serum concentrations varied
among patients and did not correlate with the dose administered. Romiplostim serum levels appear
inversely related to platelet counts.
Distribution
The volume of distribution of romiplostim following intravenous administration of romiplostim
decreased nonlinearly from 122, 78.8, to 48.2 ml/kg for intravenous doses of 0.3, 1.0 and 10 μg/kg,
respectively in healthy subjects. This non-linear decrease in volume of distribution is in line with the
(megakaryocyte and platelet) target-mediated binding of romiplostim, which may be saturated at the
higher doses applied.
Elimination
Elimination half-life of romiplostim in ITP patients ranged from 1 to 34 days (median, 3.5 days).
The elimination of serum romiplostim is in part dependent on the TPO receptor on platelets. As a
result for a given dose, patients with high platelet counts are associated with low serum concentrations
and
vice versa
. In another ITP clinical study, no accumulation in serum concentrations was observed
after 6 weekly doses of romiplostim (3 μg/kg).
Special patient populations
Pharmacokinetics of romiplostim in patients with renal and hepatic impairment has not been
investigated. Romiplostim pharmacokinetics appear not affected by age, weight and gender to a
clinically significant extent.
5.3 Preclinical safety data
Multiple dose romiplostim toxicology studies were conducted in rats for 4 weeks and in monkeys for
up to 6 months. In general, effects observed during these studies were related to the thrombopoietic
activity of romiplostim and were similar regardless of study duration. Injection site reactions were also
related to romiplostim administration. Myelofibrosis has been observed in the bone marrow of rats at
all tested dose levels. In these studies, myelofibrosis was not observed in animals after a 4-week post-
treatment recovery period, indicating reversibility.
In 1-month rat and monkey toxicology studies, a mild decrease in red blood cell count, haematocrit
and haemoglobin was observed. There was also a stimulatory effect on leukocyte production, as
peripheral blood counts for neutrophils, lymphocytes, monocytes, and eosinophils were mildly
increased. In the longer duration chronic monkey study, there was no effect on the erythroid and
leukocytic lineages when romiplostim was administered for 6 months where the administration of
romiplostim was decreased from thrice weekly to once weekly. Additionally, in the phase 3 pivotal
studies, romiplostim did not affect the red blood cell and white blood cells lineages relative to placebo
treated subjects.
Due to the formation of neutralising antibodies pharmacodynamic effects of romiplostim in rats were
often decreasing at prolonged duration of administration. Toxicokinetic studies showed no interaction
of the antibodies with the measured concentrations. Although high doses were tested in the animal
studies, due to differences between the laboratory species and humans with regard to the sensitivity for
27
the pharmacodynamic effect of romiplostim and the effect of neutralising antibodies, safety margins
cannot be reliably estimated.
Carcinogenesis:
The carcinogenic potential of romiplostim has not been evaluated. Therefore, the risk
of potential carcinogenicity of romiplostim in humans remains unknown.
Reproductive toxicology:
In all developmental studies neutralising antibodies were formed, which may
have inhibited romiplostim effects. In embryo-foetal development studies in mice and rats, reductions
in maternal body weight were found only in mice. In mice there was evidence of increased post-
implantation loss. In a prenatal and postnatal development study in rats an increase of the duration of
gestation and a slight increase in the incidence of peri-natal pup mortality was found. Romiplostim is
known to cross the placental barrier in rats and may be transmitted from the mother to the developing
foetus and stimulate foetal platelet production. Romiplostim had no observed effect on the fertility of
rats.
6.
6.1 List of excipients
Mannitol (E421)
Sucrose
L-histidine
Hydrochloric acid (for pH adjustment)
Polysorbate 20
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products, except those mentioned in section 6.6.
6.3 Shelf life
3 years.
After reconstitution: Chemical and physical in-use stability has been demonstrated for 24 hours at
25°C and for 24 hours at 2°C – 8°C, when protected from light and kept in the original vial.
From a microbiological point of view, the product should be used immediately. If not used
immediately, in-use storage times and conditions prior to use are the responsibility of the user and
would normally not be longer than 24 hours at 25°C or 24 hours in a refrigerator (2°C – 8°C),
protected from light.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C).
Do not freeze.
Store in the original carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
6.5 Nature and contents of container
5 ml vial (type 1 clear glass) with a stopper (chlorobutyl rubber), seal (aluminium) and a flip-off cap
(polypropylene).
Carton containing 1 or 4 vials of 500 µg of romiplostim.
28
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
Nplate is a sterile but unpreserved product and is intended for single use only. Nplate should be
reconstituted in accordance with good aseptic practice.
Nplate 500 micrograms powder for solution for injection should be reconstituted with 1.2 ml sterile
water for injections, yielding a deliverable volume of 1 ml. An additional overfill is included in each
vial to ensure that 500 µg of romiplostim can be delivered.
Sodium chloride solutions or bacteriostatic water should not be used when reconstituting the medicinal
product.
Water for injections should be injected into the vial. The vial contents may be swirled gently and
inverted during dissolution.
The vial should not be shaken or vigorously agitated.
Generally,
dissolution of Nplate takes less than 2 minutes. Visually inspect the solution for particulate matter and
discolouration before administration. The reconstituted solution should be clear and colourless and
should not be administered if particulate matter and/or discolouration are observed.
For the storage condition of the reconstituted product see section 6.3.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
The Netherlands
8.
EU/1/08/497/002
EU/1/08/497/004
9.
4 February 2009
Detailed information on this product is available on the website of the European Medicines Agency
29
1.
Nplate 250 micrograms powder and solvent for solution for injection
2.
Each vial contains 250 µg of romiplostim. After reconstitution, a deliverable volume of 0.5 ml
solution contains 250 µg of romiplostim (500 µg/ml). An additional overfill is included in each vial to
ensure that 250 µg of romiplostim can be delivered.
Romiplostim is produced by recombinant DNA technology in
Escherichia coli
(
E. coli
).
For a full list of excipients, see section 6.1.
3.
Powder and solvent for solution for injection.
The powder is white.
The solvent is a clear colourless liquid.
4.
Nplate is indicated for adult chronic immune (idiopathic) thrombocytopenic purpura (ITP)
splenectomised patients who are refractory to other treatments (e.g. corticosteroids, immunoglobulins).
Nplate may be considered as second line treatment for adult non-splenectomised patients where
surgery is contra-indicated.
Treatment should remain under the supervision of a physician who is experienced in the treatment of
haematological diseases.
Posology
Nplate should be administered once weekly as a subcutaneous injection.
Initial dose
The initial dose of romiplostim is 1 µg/kg based on actual body weight.
Dose calculation
Weight* in kg x Dose in µg/kg = Individual patient dose in µg
Volume to administer: Dose in µg x
1 ml
500 µg
= Amount to inject in ml
30
Initial or subsequent
once weekly dose:





Example:
75 kg patient is initiated at 1 µg/kg of romiplostim.
The individual patient dose =
75 kg x 1 µg = 75 µg
The corresponding amount of Nplate solution to inject =
75 µg x
1 ml
= 0.15 ml
500 µg
*Actual body weight at initiation of treatment should always be used when calculating dose of
romiplostim. Future dose adjustments are based on changes in platelet counts only and made in
1 µg/kg increments (see table below).
Dose adjustments
A subject’s actual body weight at initiation of therapy should be used to calculate dose. The once
weekly dose of romiplostim should be increased by increments of 1 μg/kg until the patient achieves a
platelet count ≥ 50 x 10
9
/l. Platelet counts should be assessed weekly until a stable platelet count
(≥ 50 x 10
9
/l for at least 4 weeks without dose adjustment) has been achieved. Platelet counts should
be assessed monthly thereafter. Do not exceed a maximum once weekly dose of 10 μg/kg.
Adjust the dose as follows:
Platelet count
(x 10
9
/l)
Action
< 50
Increase once weekly dose by 1 μg/kg
consecutive weeks
Decrease once weekly dose by 1 μg/kg
Do not administer, continue to assess the platelet count weekly
> 250
After the platelet count has fallen to < 150 x 10
9
/l, resume dosing with once
weekly dose reduced by 1 μg/kg
Due to the interindividual variable platelet response, in some patients platelet count may abruptly fall
below 50 x 10
9
/l after dose reduction or treatment discontinuation. In these cases, if clinically
appropriate, higher cut-off levels of platelet count for dose reduction (200 x 10
9
/l) and treatment
interruption (400 x 10
9
/l) may be considered according to medical judgement.
A loss of response or failure to maintain a platelet response with romiplostim within the recommended
dosing range should prompt a search for causative factors (see section 4.4, loss of response to
romiplostim).
Treatment discontinuation
Treatment with romiplostim should be discontinued if the platelet count does not increase to a level
sufficient to avoid clinically important bleeding after four weeks of romiplostim therapy at the highest
weekly dose of 10 μg/kg.
Patients should be clinically evaluated periodically and continuation of treatment should be decided on
an individual basis by the treating physician. The reoccurrence of thrombocytopenia is likely upon
discontinuation of treatment (see section 4.4).
Method of administration
For subcutaneous use.
After reconstitution of the powder, Nplate solution for injection is administered subcutaneously. The
injection volume may be very small. A syringe with graduations of 0.01 ml should be used.
31
> 150 for two












For instructions on reconstitution of Nplate before administration, see section 6.6.
Elderly patients (≥ 65 years)
No overall differences in safety or efficacy have been observed in patients < 65 and ≥ 65 years of age
(see section 5.1). Although based on these data no adjustment of the dosing regimen is required for
older patients, care is advised considering the small number of elderly patients included in the clinical
trials so far.
Paediatric population
Nplate is not recommended for use in children below age 18 due to insufficient data on safety or
efficacy. No recommendation on a posology can be made in this population.
Hepatic Impairment
Romiplostim should not be used in patients with moderate to severe hepatic impairment (Child-Pugh
score ≥ 7) unless the expected benefit outweighs the identified risk of portal venous thrombosis in
patients with thrombocytopenia associated to hepatic insufficiency treated with TPO agonists (see
section 4.4).
If the use of romiplostim is deemed necessary, platelet count should be closely monitored to minimise
the risk of thromboembolic complications.
Renal impairment
No formal clinical studies have been conducted in these patient populations. Nplate should be used
with caution in these populations.
Hypersensitivity to the active substance, to any of the excipients or to
E. coli
derived proteins.
The following special warnings and precautions have been actually observed or are potential class
effects based on the pharmacological mechanism of action of thrombopoietin (TPO) receptor
stimulators.
Reoccurrence of thrombocytopenia and bleeding after cessation of treatment
Thrombocytopenia is likely to reoccur upon discontinuation of treatment with romiplostim. There is an
increased risk of bleeding if romiplostim treatment is discontinued in the presence of anticoagulants or
anti-platelet agents. Patients should be closely monitored for a decrease in platelet count and medically
managed to avoid bleeding upon discontinuation of treatment with romiplostim. It is recommended
that, if treatment with romiplostim is discontinued, ITP treatment be restarted according to current
treatment guidelines. Additional medical management may include cessation of anticoagulant and/or
antiplatelet therapy, reversal of anticoagulation, or platelet support.
Increased bone marrow reticulin
Increased bone marrow reticulin is believed to be a result of TPO receptor stimulation, leading to an
increased number of megakaryocytes in the bone marrow, which may subsequently release cytokines.
Increased reticulin may be suggested by morphological changes in the peripheral blood cells and can
be detected through bone marrow biopsy. Therefore, examinations for cellular morphological
abnormalities using peripheral blood smear and complete blood count (CBC) prior to and during
32
treatment with romiplostim are recommended. See section 4.8 for information on the increases of
reticulin observed in romiplostim clinical trials.
If a loss of efficacy and abnormal peripheral blood smear is observed in patients, administration of
romiplostim should be discontinued, a physical examination should be performed, and a bone marrow
biopsy with appropriate staining for reticulin should be considered. If available, comparison to a prior
bone marrow biopsy should be made. If efficacy is maintained and abnormal peripheral blood smear is
observed in patients, the physician should follow appropriate clinical judgment, including
consideration of a bone marrow biopsy, and the risk-benefit of romiplostim and alternative ITP
treatment options should be re-assessed.
Thrombotic/thromboembolic complications
Platelet counts above the normal range present a theoretical risk for thrombotic/thromboembolic
complications. The incidence of thrombotic/thromboembolic events observed in clinical trials was
similar between romiplostim and placebo, and an association between these events and elevated
platelet counts was not observed. Caution should be used when administering romiplostim to patients
with known risk factors for thromboembolism including but not limited to inherited (e.g. Factor V
Leiden) or acquired risk factors (e.g. ATIII deficiency, antiphospholipid syndrome), advanced age,
patients with prolonged periods of immobilisation, malignancies, contraceptives and hormone
replacement therapy, surgery/trauma, obesity and smoking.
Cases of thromboembolic events (TEEs), including portal vein thrombosis, have been reported in
patients with chronic liver disease receiving romiplostim. Romiplostim should be used with caution in
these populations. Dose adjustment guidelines should be followed (see section 4.2).
Progression of existing haematopoietic malignancies or Myelodysplastic Syndromes (MDS)
TPO receptor stimulators are growth factors that lead to thrombopoietic progenitor cell expansion,
differentiation, and platelet production. The TPO receptor is predominantly expressed on the surface
of cells of the myeloid lineage. For TPO receptor stimulators there is a theoretical concern that they
may stimulate the progression of existing haematopoietic malignancies or MDS.
The diagnosis of ITP in adults and elderly patients should have been confirmed by the exclusion of
other clinical entities presenting with thrombocytopenia. Consideration should be given to performing
a bone marrow aspirate and biopsy over the course of the disease and treatment, particularly in
patients over 60 years of age, those with systemic symptoms or abnormal signs.
Romiplostim should not be used for the treatment of thrombocytopenia due to MDS or any other cause
of thrombocytopenia other than ITP outside of clinical trials. The risk-benefit profile for romiplostim
has not been established in MDS or other non-ITP patient populations. In clinical studies of treatment
with romiplostim in patients with MDS, there were reported cases of progression to acute myeloid
leukaemia (AML), however this is an expected clinical outcome of MDS and the relationship to
romiplostim treatment is unclear.
Loss of response to romiplostim
A loss of response or failure to maintain a platelet response with romiplostim treatment within the
recommended dosing range should prompt a search for causative factors, including immunogenicity
(see section 4.8) and increased bone marrow reticulin (see above).
Effects of romiplostim on red and white blood cells
Alterations in red (decrease) and white (increase) blood cell parameters have been observed in non-
clinical toxicology studies (rat and monkey) but not in ITP patients. Monitoring of these parameters
should be considered in patients treated with romiplostim.
33
No interaction studies have been performed. The potential interactions of romiplostim with co-
administered medicinal products due to binding to plasma proteins remain unknown.
Medicinal products used in the treatment of ITP in combination with romiplostim in clinical studies
included corticosteroids, danazol, and/or azathioprine, intravenous immunoglobulin (IVIG), and
anti-D immunoglobulin. Platelet counts should be monitored when combining romiplostim with other
medicinal products for the treatment of ITP in order to avoid platelet counts outside of the
recommended range (see section 4.2).
Corticosteroids, danazol, and azathioprine use may be reduced or discontinued when given in
combination with romiplostim (see section 5.1). Platelet counts should be monitored when reducing or
discontinuing other ITP treatments in order to avoid platelet counts below the recommended range
(see section 4.2).
Pregnancy
For romiplostim no clinical data on exposed pregnancies are available.
Studies in animals have
shown reproductive toxicity, such as transplacental passage and increased
foetal platelet counts in rats (see section 5.3). The potential risk for humans is unknown.
Romiplostim should not be used during pregnancy unless clearly necessary.
Breast-feeding
There are no data on excretion of romiplostim in human milk. However, excretion is likely and a risk
to the suckling child cannot be excluded. A decision on whether to continue/discontinue breast-feeding
or to continue/discontinue therapy with romiplostim should be made taking into account the benefit of
breast-feeding to the child and the benefit of romiplostim therapy to the woman.
No studies on the effects on the ability to drive and use machines have been performed. However,
patients should be informed that in clinical trials mild to moderate, transient bouts of dizziness were
experienced by some patients, which may affect the ability to drive or use machines.
a.
Summary of the safety profile
Based on an analysis of all adult ITP patients receiving romiplostim in 4 controlled and 5 uncontrolled
clinical studies, the overall subject incidence of all adverse reactions for romiplostim-treated subjects
was 91.5% (248/271). The mean duration of exposure to romiplostim in this study population was
50 weeks.
b.
Tabulated list of adverse reactions
Frequencies are defined as: Very common (≥ 1/10), Common (≥ 1/100 to < 1/10) and Uncommon
(≥ 1/1000 to < 1/100). Within each MedDRA system organ class and frequency grouping, undesirable
effects are presented in order of decreasing incidence.
MedDRA system
organ class
Very common Common
Uncommon
34






MedDRA system
organ class
Very common Common
Uncommon
Blood and lymphatic
system disorders
Bone marrow
disorder*
Thrombocytopenia*
Anaemia
Aplastic anaemia
Bone marrow failure
Leukocytosis
Splenomegaly
Thrombocythaemia
Platelet count increased
Platelet count abnormal
Cardiac disorders
Myocardial infarction
Heart rate increased
Ear and labyrinth
disorders
Vertigo
Eye disorders
Conjunctival haemorrhage
Accommodation disorder
Blindness
Eye disorder
Eye pruritus
Lacrimation increased
Papilloedema
Visual disturbances
Gastrointestinal
disorders
Nausea
Diarrhoea
Abdominal pain
Constipation
Dyspepsia
Vomiting
Rectal haemorrhage
Breath odour
Dysphagia
Gastro-oesophageal reflux
disease
Haematochezia
Mouth haemorrhage
Stomach discomfort
Stomatitis
Tooth discolouration
General disorders and
administration site
conditions
Fatigue
Oedema peripheral
Influenza like illness
Pain
Asthenia
Pyrexia
Chills
Injection site reaction
Injection site haemorrhage
Chest pain
Irritability
Malaise
Face oedema
Feeling hot
Feeling jittery
Hepatobiliary
disorders
Portal vein thrombosis
Increase in transaminase
Infections and
infestations
Influenza
Localised infection
Nasopharyngitis
Injury, poisoning and
procedural
complications
Contusion
Investigations
Blood pressure increased
Blood lactate dehydrogenase
increased
Body temperature increased
Weight decreased
Weight increased
35
















MedDRA system
organ class
Very common Common
Uncommon
Metabolism and
nutrition disorders
Alcohol intolerance
Anorexia
Decreased appetite
Dehydration
Gout
Musculoskeletal and
connective tissue
disorders
Arthralgia
Myalgia
Muscle spasms
Pain in extremity
Back pain
Bone pain
Muscle tightness
Muscular weakness
Shoulder pain
Muscle twitching
Neoplasms benign,
malignant and
unspecified (incl cysts
and polyps)
Multiple myeloma
Myelofibrosis
Nervous system
disorders
Headache Dizziness
Migraine
Paraesthesia
Clonus
Dysgeusia
Hypoaesthesia
Hypogeusia
Neuropathy peripheral
Transverse sinus thrombosis
Psychiatric disorders
Insomnia
Depression
Abnormal dreams
Renal and urinary
disorders
Protein urine present
Reproductive system
and breast disorders
Vaginal haemorrhage
Respiratory, thoracic
and mediastinal
disorders
Pulmonary embolism* Cough
Rhinorrhoea
Dry throat
Dyspnoea
Nasal congestion
Painful respiration
Skin and subcutaneous
tissue disorders
Pruritus
Ecchymosis
Rash
Alopecia
Photosensitivity reaction
Acne
Dermatitis contact
Dry skin
Eczema
Erythema
Exfoliative rash
Hair growth abnormal
Prurigo
Purpura
Rash papular
Rash pruritic
Skin nodule
Skin odour abnormal
Urticaria
36















MedDRA system
organ class
Very common Common
Uncommon
Vascular disorders
Flushing
Deep vein thrombosis
Hypotension
Peripheral embolism
Peripheral ischaemia
Phlebitis
Thrombophlebitis superficial
Thrombosis
* see section 4.4
c.
Description of selected adverse reactions
In addition the reactions listed below have been deemed to be related to romiplostim treatment.
Thrombocytosis
Based on an analysis of all adult ITP patients receiving romiplostim in 4 controlled and 5 uncontrolled
clinical studies, 3 events of thrombocytosis were reported, n = 271.
No clinical sequelae were reported
in association with the elevated platelet counts in any of the 3 subjects.
Thrombocytopenia after cessation of treatment
Based on an analysis of all adult ITP patients receiving romiplostim in 4 controlled and 5 uncontrolled
clinical studies, 4 events of thrombocytopenia after cessation of treatment were reported, n = 271 (see
section 4.4).
Increased bone marrow reticulin
In clinical studies, romiplostim treatment was discontinued in 4 of the 271 patients because of bone
marrow reticulin deposition. In 6 additional patients reticulin was observed upon bone marrow biopsy
(see section 4.4).
Immunogenicity
Clinical studies in adult ITP patients examined antibodies to romiplostim.
While 5.8% and 3.9% of the subjects were positive for developing binding antibodies to romiplostim
and TPO respectively, only 2 subjects (0.4%) were positive for neutralizing antibodies to romiplostim
but these antibodies did not cross react with endogenous TPO. Both subjects tested negative for
neutralising antibodies to romiplostim at 4 months after the end of dosing. The incidence of pre-
existing antibodies to romiplostim and TPO was 8.0% and 5.4%, respectively.
As with all therapeutic proteins, there is a potential for immunogenicity. If formation of neutralising
antibodies is suspected, contact the local representative of the Marketing Authorisation Holder (see
section 6 of the Package Leaflet) for antibody testing.
Adverse reactions from spontaneous reporting:
The frequency category of the adverse reactions identified from spontaneous reporting that have not
been reported in clinical trials cannot be estimated (Frequency: not known). The adverse reactions
identified from spontaneous reporting include:
Vascular disorders: Erythromelalgia.
37







No adverse effects were seen in rats given a single dose of 1000 μg/kg or in monkeys after repeated
administration of romiplostim at 500 µg/kg (100 or 50 times the maximum clinical dose of 10 µg/kg,
respectively).
In the event of overdose, platelet counts may increase excessively and result in
thrombotic/thromboembolic complications. If the platelet counts are excessively increased,
discontinue Nplate and monitor platelet counts. Reinitiate treatment with Nplate in accordance with
dosing and administration recommendations (see section 4.2).
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Antihemorrhagics, ATC code: B02BX04
Romiplostim is an Fc-peptide fusion protein (peptibody) that signals and activates intracellular
transcriptional pathways via the thrombopoietin (TPO) receptor (also known as cMpl) to increase
platelet production. The peptibody molecule is comprised of a human immunoglobulin IgG1 Fc
domain, with each single-chain subunit covalently linked at the C-terminus to a peptide chain
containing 2 TPO receptor-binding domains.
Romiplostim has no amino acid sequence homology to endogenous TPO. In pre-clinical and clinical
studies no anti-romiplostim antibodies cross reacted with endogenous TPO.
Clinical data
The safety and efficacy of romiplostim have been evaluated for up to 3 years of continuous treatment.
In clinical studies, treatment with romiplostim resulted in dose-dependent increases in platelet count.
Time to reach the maximum effect on platelet count is approximately 10-14 days, and is independent
of the dose. After a single subcutaneous dose of 1 to 10 µg/kg romiplostim in ITP patients, the peak
platelet count was 1.3 to 14.9 times greater than the baseline platelet count over a 2 to 3 week period
and the response was variable among patients. The platelet counts of ITP patients who received
6 weekly doses of 1 or 3 µg/kg of romiplostim were within the range of 50 to 450 x 10
9
/l for most
patients. Of the 271 patients who received romiplostim in ITP clinical studies, 55 (20%) were age 65
and over, and 27 (10%) were 75 and over. No overall differences in safety or efficacy have been
observed between older and younger patients in the placebo-controlled studies.
Results from pivotal placebo-controlled studies
The safety and efficacy of romiplostim was evaluated in two placebo-controlled, double-blind studies
in adults with ITP who had completed at least one treatment prior to study entry and are representative
of the entire spectrum of such ITP patients.
Study S1 (212) evaluated patients who were non-splenectomised and had an inadequate response or
were intolerant to prior therapies. Patients had been diagnosed with ITP for approximately 2 years at
the time of study entry. Patients had a median of 3 (range, 1 to 7) treatments for ITP prior to study
entry. Prior treatments included corticosteroids (90% of all patients), immunoglobulins (76%),
rituximab (29%), cytotoxic therapies (21%), danazol (11%), and azathioprine (5%). Patients had a
median platelet count of 19 x 10
9
/l at study entry.
Study S2 (105) evaluated patients who were splenectomised and continued to have thrombocytopenia.
Patients had been diagnosed with ITP for approximately 8 years at the time of study entry. In addition
to a splenectomy, patients had a median of 6 (range, 3 to 10) treatments for ITP prior to study entry.
38
Prior treatments included corticosteroids (98% of all patients), immunoglobulins (97%), rituximab
(71%), danazol (37%), cytotoxic therapies (68%), and azathioprine (24%). Patients had a median
platelet count of 14 x 10
9
/l at study entry.
Both studies were similarly designed. Patients (≥ 18 years) were randomised in a 2:1 ratio to receive a
starting dose of romiplostim 1 µg/kg or placebo. Patients received single subcutaneous weekly
injections for 24 weeks. Doses were adjusted to maintain (50 to 200 x 10
9
/l) platelet counts. In both
studies, efficacy was determined by an increase in the proportion of patients who achieved a durable
platelet response. The median average weekly dose for splenectomised patients was 3 µg/kg and for
non-splenectomised patients was 2 µg/kg.
A significantly higher proportion of patients receiving romiplostim achieved a durable platelet
response compared to patients receiving placebo in both studies. Following the first 4-weeks of study
romiplostim maintained platelet counts ≥ 50 x 10
9
/l in between 50% to 70% of patients during the
6 month treatment period in the placebo-controlled studies. In the placebo group, 0% to 7% of patients
were able achieve a platelet count response during the 6 months of treatment. A summary of the key
efficacy endpoints is presented below.
Summary of key efficacy results from placebo-controlled studies
Study 1
non-splenectomised
patients
Study 2
splenectomised patients
Combined
studies 1 & 2
romiplostim
(n = 41)
Placebo
(n = 21)
romiplostim
(n = 42)
Placebo
(n = 21)
romiplostim
(n = 83)
Placebo
(n = 42)
No. (%)
patients
with
durable
platelet
response
a
25 (61%) 1 (5%) 16 (38%) 0 (0%) 41 (50%) 1 (2%)
(95%
CI)
(45%, 76%) (0%, 24%) (24%, 54%) (0%, 16%) (38%, 61%) (0%, 13%)
p-
value
< 0.0001
0.0013
< 0.0001
No. (%)
patients
with
overall
platelet
response
b
36 (88%) 3 (14%) 33 (79%) 0 (0%) 69 (83%) 3 (7%)
(95%
CI)
(74%, 96%) (3%, 36%) (63%, 90%) (0%, 16%) (73%, 91%) (2%, 20%)
p-
value
< 0.0001
< 0.0001
< 0.0001
39
















Study 1
non-splenectomised
patients
Study 2
splenectomised patients
Combined
studies 1 & 2
romiplostim
(n = 41)
Placebo
(n = 21)
romiplostim
(n = 42)
Placebo
(n = 21)
romiplostim
(n = 83)
Placebo
(n = 42)
Mean no.
weeks with
platelet
response
c
15
1
12
0
14
1
(SD)
3.5
7.5
7.9
0.5
7.8
2.5
p-
value
< 0.0001
< 0.0001
< 0.0001
No. (%)
patients
requiring
rescue
therapies
d
8(20%) 13 (62%) 11 (26%) 12 (57%) 19 (23%) 25 (60%)
(95%
CI)
(9%, 35%) (38%, 82%) (14%, 42%) (34%, 78%) (14%, 33%) (43%, 74%)
p-
value
0.001
0.0175
< 0.0001
No. (%)
patients
with
durable
platelet
response
with stable
dose
e
21 (51%) 0 (0%) 13 (31%) 0 (0%) 34 (41%) 0 (0%)
(95%
CI)
(35%, 67%) (0%, 16%) (18%, 47%) (0%, 16%) (30%, 52%) (0%, 8%)
0.0001 0.0046 < 0.0001
a
Durable platelet response was defined as weekly platelet count ≥ 50 x 10
9
/l for 6 or more times for study
weeks 18-25 in the absence of rescue therapies any time during the treatment period.
b
Overall platelet response is defined as achieving durable or transient platelet responses. Transient platelet
response was defined as weekly platelet count≥ 50 x 10
9
/l for 4 or more times during study weeks 2-25
but wi thout durable p latelet r esponse. P atient may n ot h ave a weekly r esponse wi thin 8 weeks af ter
receiving any rescue medicinal products.
c
Number of weeks with platelet response is defined as number of weeks with platelet counts ≥ 50 x 10
9
/l
during study weeks 2-25. Patient may not have a weekly response within 8 weeks after receiving any
rescue medicinal products.
d
Rescue therapies defined as any therapy administered to raise platelet counts. Patients requiring rescue
medicinal products were not considered for durable platelet response. Rescue therapies allowed in the
study were IVIG, platelet transfusions, anti-D immunoglobulin, and corticosteroids.
e
Stable dose defined as dose maintained within ± 1 µg/kg during the last 8 weeks of treatment.
Reduction in permitted concurrent ITP medical therapies
In both placebo-controlled, double-blind studies, patients already receiving ITP medical therapies at a
constant dosing schedule were allowed to continue receiving these medical treatments throughout the
study (corticosteroids, danazol and/or azathioprine). Twenty-one non-splenectomised and
18 splenectomised patients received on-study ITP medical treatments (primarily corticosteroids) at the
start of study. All (100%) splenectomised patients who were receiving romiplostim were able to
reduce the dose by more than 25% or discontinue the concurrent ITP medical therapies by the end of
the treatment period compared to 17% of placebo treated patients. Seventy-three percent of
non-splenectomised patients receiving romiplostim were able to reduce the dose by more than 25% or
40
p-
value



















discontinue concurrent ITP medical therapies by the end of the study compared to 50% of placebo
treated patients (see section 4.5).
Bleeding events
Across the entire ITP clinical programme an inverse relationship between bleeding events and platelet
counts was observed. All clinically significant (≥ grade 3) bleeding events occurred at platelet counts
< 30 x 10
9
/l. All bleeding events ≥ grade 2 occurred at platelet counts < 50 x 10
9
/l. No statistically
significant differences in the overall incidence of bleeding events were observed between Nplate and
placebo treated patients.
In the two placebo-controlled studies, 9 patients reported a bleeding event that was considered serious
(5 [6.0%] romiplostim, 4 [9.8%] placebo; Odds Ratio [romiplostim/placebo] = 0.59; 95% CI = (0.15,
2.31)). Bleeding events that were grade 2 or higher were reported by 15% of patients treated with
romiplostim and 34% of patients treated with placebo (Odds Ratio; [romiplostim/placebo] = 0.35; 95%
CI = (0.14, 0.85)).
5.2 Pharmacokinetic properties
The pharmacokinetics of romiplostim involved target-mediated disposition, which is presumably
mediated by TPO receptors on platelets and other cells of the thrombopoietic lineage such as
megakaryocytes.
Absorption
After subcutaneous administration of 3 to 15 μg/kg romiplostim, maximum romiplostim serum levels
in ITP patients were obtained after 7-50 hours (median 14 hours). The serum concentrations varied
among patients and did not correlate with the dose administered. Romiplostim serum levels appear
inversely related to platelet counts.
Distribution
The volume of distribution of romiplostim following intravenous administration of romiplostim
decreased nonlinearly from 122, 78.8, to 48.2 ml/kg for intravenous doses of 0.3, 1.0 and 10 μg/kg,
respectively in healthy subjects. This non-linear decrease in volume of distribution is in line with the
(megakaryocyte and platelet) target-mediated binding of romiplostim, which may be saturated at the
higher doses applied.
Elimination
Elimination half-life of romiplostim in ITP patients ranged from 1 to 34 days (median, 3.5 days).
The elimination of serum romiplostim is in part dependent on the TPO receptor on platelets. As a
result for a given dose, patients with high platelet counts are associated with low serum concentrations
and
vice versa
. In another ITP clinical study, no accumulation in serum concentrations was observed
after 6 weekly doses of romiplostim (3 μg/kg).
Special patient populations
Pharmacokinetics of romiplostim in patients with renal and hepatic impairment has not been
investigated. Romiplostim pharmacokinetics appear not affected by age, weight and gender to a
clinically significant extent.
5.3 Preclinical safety data
Multiple dose romiplostim toxicology studies were conducted in rats for 4 weeks and in monkeys for
up to 6 months. In general, effects observed during these studies were related to the thrombopoietic
activity of romiplostim and were similar regardless of study duration. Injection site reactions were also
41
related to romiplostim administration. Myelofibrosis has been observed in the bone marrow of rats at
all tested dose levels. In these studies, myelofibrosis was not observed in animals after a 4-week post-
treatment recovery period, indicating reversibility.
In 1-month rat and monkey toxicology studies, a mild decrease in red blood cell count, haematocrit
and haemoglobin was observed. There was also a stimulatory effect on leukocyte production, as
peripheral blood counts for neutrophils, lymphocytes, monocytes, and eosinophils were mildly
increased. In the longer duration chronic monkey study, there was no effect on the erythroid and
leukocytic lineages when romiplostim was administered for 6 months where the administration of
romiplostim was decreased from thrice weekly to once weekly. Additionally, in the phase 3 pivotal
studies, romiplostim did not affect the red blood cell and white blood cells lineages relative to placebo
treated subjects.
Due to the formation of neutralising antibodies pharmacodynamic effects of romiplostim in rats were
often decreasing at prolonged duration of administration. Toxicokinetic studies showed no interaction
of the antibodies with the measured concentrations. Although high doses were tested in the animal
studies, due to differences between the laboratory species and humans with regard to the sensitivity for
the pharmacodynamic effect of romiplostim and the effect of neutralising antibodies, safety margins
cannot be reliably estimated.
Carcinogenesis:
The carcinogenic potential of romiplostim has not been evaluated. Therefore, the risk
of potential carcinogenicity of romiplostim in humans remains unknown.
Reproductive toxicology:
In all developmental studies neutralising antibodies were formed, which may
have inhibited romiplostim effects. In embryo-foetal development studies in mice and rats, reductions
in maternal body weight were found only in mice. In mice there was evidence of increased post-
implantation loss. In a prenatal and postnatal development study in rats an increase of the duration of
gestation and a slight increase in the incidence of peri-natal pup mortality was found. Romiplostim is
known to cross the placental barrier in rats and may be transmitted from the mother to the developing
foetus and stimulate foetal platelet production. Romiplostim had no observed effect on the fertility of
rats.
6.
6.1 List of excipients
Mannitol (E421)
Sucrose
L-histidine
Hydrochloric acid (for pH adjustment)
Polysorbate 20
Solvent:
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products, except those mentioned in section 6.6.
6.3 Shelf life
2 years.
After reconstitution: Chemical and physical in-use stability has been demonstrated for 24 hours at
25°C and for 24 hours at 2°C – 8°C, when protected from light and kept in the original vial.
42
From a microbiological point of view, the product should be used immediately. If not used
immediately, in-use storage times and conditions prior to use are the responsibility of the user and
would normally not be longer than 24 hours at 25°C or 24 hours in a refrigerator (2°C – 8°C),
protected from light.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C).
Do not freeze.
Store in the original carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
6.5 Nature and contents of container
Powder:
5 ml vial (type 1 clear glass) with a stopper (chlorobutyl rubber), seal (aluminium) and a flip-off cap
(polypropylene).
Solvent:
Pre-filled syringe (type 1 clear glass with bromobutyl rubber plunger) containing 0.72 ml of water for
injections for reconstitution.
Pack size:
Nplate is supplied as a 1 pack or multipack comprising 4 packs. Each pack contains:
1 vial of 250 micrograms romiplostim.
1 pre-filled syringe containing 0.72 ml of water for injections for reconstitution.
1 plunger rod for the pre-filled syringe.
1 sterile vial adapter.
1 sterile 1 ml
Luer lock syringe.
1 sterile safety needle.
4 alcohol swabs.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
Nplate is a sterile but unpreserved product and is intended for single use only. Nplate should be
reconstituted in accordance with good aseptic practice.
For the storage condition of the reconstituted product see section 6.3.
•
Nplate 250 micrograms powder for solution for injection
should be reconstituted with
0.72 ml sterile water for injections, yielding a deliverable volume of 0.5 ml. An additional
overfill is included in each vial to ensure that 250 µg of romiplostim can be delivered.
or
•
Nplate 500 micrograms powder for solution for injection
should be reconstituted with 1.2 ml
sterile water for injections, yielding a deliverable volume of 1 ml. An additional overfill is
included in each vial to ensure that 500 µg of romiplostim can be delivered.
From a microbiological point of view, the product should be used immediately. If not used
immediately, in-use storage times and conditions prior to use are the responsibility of the user and
43
would normally not be longer than 24 hours at 25°C or 24 hours in a refrigerator (2°C – 8°C),
protected from light.
1.
Remove the plastic cap from Nplate powder vial and
clean rubber stopper using the provided alcohol swab.
2.
Attach vial adapter to Nplate vial by peeling off
paper
backing from vial adapter, keeping the vial adapter in
its packaging.
Keeping the
vial
Note: To prevent contamination of the product, do
not touch the vial adapter spike or Luer lock.
3.
Remove and discard vial adapter packaging.
4.
Attach plunger rod to the pre-filled syringe of water
for injections
by twisting the plunger rod clockwise onto
the syringe plunger, until you feel a slight resistance.
5.
Holding the pre-filled syringe of water for injections
with one hand, bend the tip of the white plastic cover
downward with your other hand. This will break the
seal of the white plastic cover. Once the seal is
broken, pull cover off to separate the grey rubber cap
from the clear plastic syringe tip.
6.
Keeping the vial on the bench, attach the pre-filled
syringe of water for injections to vial adapter:
hold the
outer edge of the vial adapter with one hand and twist the
syringe tip clockwise onto the adapter with the other hand
until you feel a slight resistance.
into powder
vial. Water should flow slowly onto powder. GENTLY
swirl the vial until all of the powder has dissolved and the
fluid in the vial is clear and colourless.
Do not
shake or agitate vial
NOTE:
This may take up to
2 minutes for the powder to
completely dissolve.
Note: From a microbiological point of view, the
product must be used immediately after
reconstitution. If reconstituted product is not used
immediately, the syringe should not be removed from
the vial adapter to maintain microbiological integrity.
44
on the bench,
push the
vial adapter down onto the centre of the vial until it is
firmly in place.
7.
Very slowly and gently expel all water














Before continuing:
Do
visually inspect the reconstituted solution for particulate matter and/or discoloration. The
reconstituted solution should be clear and colourless and should not be administered if particulate
matter and/or discolouration are observed.
Do
make sure solution is fully dissolved before removing syringe.
8.
Remove the empty pre-filled syringe from the vial
adapter.
9.
Remove 1 ml administration syringe from package.
Attach the 1 ml syringe to
vial adapter of
reconstituted
solution
10.
Turn assembled syringe-vial unit upside down,
so the
vial of reconstituted product is above the syringe.
Withdraw all
of the medicinal product solution into the
administration
syringe.
11.
Ensure the correct amount of solution
for the patient
dose is in the administration
syringe by expelling any
excess solution back into the vial.
Note: Remove all air bubbles from syringe to ensure
precise solution amount is in syringe.
12.
Twist off administration syringe from vial adapter.
Attach safety needle
to the filled administration syringe
by twisting needle
clockwise
into syringe Luer lock tip.
13.
Prepare injection site with a new alcohol swab.
Pull
back on the pink safety cover
toward the syringe and
away from the needle.
Remove clear needle shield from prepared needle
by
holding syringe in one hand and carefully pulling shield
straight off with the other hand.
14.
Administer subcutaneous injection
following local
protocols and good aseptic technique.
45
by twisting the syringe tip onto the vial adapter
until you feel a slight resistance.



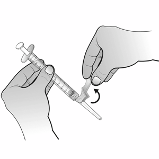











15.
After injecting, activate the pink safety cover
by
pushing the cover forward using the same hand until you
hear and/or feel it click/lock.
16.
Immediately discard syringe and needle
into an
approved Sharps Container.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
The Netherlands
8.
EU/1/08/497/005
EU/1/08/497/006
9.
4 February 2009
Detailed information on this product is available on the website of the European Medicines Agency
46
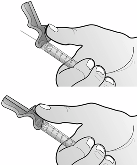





1.
FURTHER INFORMATION
What Nplate contains
- The active substance is romiplostim.
Each vial of Nplate 250 micrograms powder for solution for injection contains 250 micrograms
of romiplostim. After reconstitution, a deliverable volume of 0.5 ml solution contains
250 micrograms of romiplostim (500 micrograms/ml).
Each vial of Nplate 500 micrograms powder for solution for injection contains 500 micrograms
of romiplostim. After reconstitution, a deliverable volume of 1 ml solution contains
500 micrograms of romiplostim (500 micrograms/ml).
- The other ingredients are:
Powder:
mannitol (E421), sucrose, L-histidine, hydrochloric acid (for pH adjustment) and
polysorbate 20.
Solvent
: water for injections.
What Nplate looks like and contents of the pack
Nplate is a white powder for solution for injection supplied in a 5 ml glass vial.
Nplate is supplied as a 1 pack or multipack comprising 4 packs. Each pack contains:
1 vial of 250 micrograms or 500 micrograms of romiplostim.
1 pre-filled syringe containing 0.72 or 1.2 ml of water for injections for reconstitution.
1 plunger rod for pre-filled syringe.
1 sterile vial adapter.
1 sterile 1 ml Luer lock syringe.
1 sterile safety needle.
4 alcohol swabs.
Not all pack sizes may be marketed.
Marketing Authorisation Holder and Manufacturer
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
The Netherlands
For any information about this medicine, please contact the local representative of the Marketing
Authorisation Holder.
België/Belgique/Belgien
s.a. Amgen n.v.
Tel/Tél: +32 (0)2 7752711
Luxembourg/Luxemburg
s.a. Amgen
Belgique/Belgien
Tel/Tél: +32 (0)2 7752711
България
Амджен България ЕООД
Тел: +359 (0)2 805 7020
Magyarország
Amgen Kft.
Tel.: +36 1 35 44 700
98
Česká republika
Amgen s.r.o.
Tel: +420 2 21 773 500
Malta
Amgen B.V.
The Netherlands
Tel: +31 (0)76 5732500
Danmark
Amgen filial af Amgen AB, Sverige
Tlf: +45 39617500
Nederland
Amgen B.V.
Tel: +31 (0)76 5732500
Deutschland
AMGEN GmbH
Tel.: +49 89 1490960
Norge
Amgen AB
Tel: +47 23308000
Eesti
Amgen Switzerland AG Vilniaus filialas
Tel: +372 5125 501
Österreich
Amgen GmbH
Tel: +43 (0)1 50 217
Ελλάδα
Amgen Ελλάς Φαρμακευτικά Ε.Π.Ε.
Τηλ.: +30 210 3447000
Polska
Amgen Sp. z o.o.
Tel.: +48 22 581 3000
España
Amgen S.A.
Tel: +34 93 600 19 00
Portugal
AMGEN Biofarmacêutica, Lda.
Tel: +351 21 4220550
France
Amgen S.A.S
România
Amgen România SRL
Tel.: +4021 527 3000
Ireland
Amgen Limited
United Kingdom
Tel: +44 (0)1223 420305
Slovenija
AMGEN zdravila d.o.o.
Tel: +386 1 585 1767
Ísland
Vistor hf.
Sími: +354 535 7000
Slovenská republika
Amgen Switzerland AG Slovakia
Tel: +421 33 321 13 22
Italia
Amgen Dompé S.p.A.
Tel: +39 02 6241121
Suomi/Finland
Amgen AB, sivuliike Suomessa/Amgen AB, filial
i Finland
Puh/Tel: +358 (0)9 54900500
Kύπρος
Papaellinas & Co Ltd
Τηλ: +357 22741 741
Sverige
Amgen AB
Tel: +46 (0)8 6951100
Latvija
Amgen Switzerland AG Rīgas filiāle
Tel: +371 29284 807
United Kingdom
Amgen Limited
Tel: +44 (0)1223 420305
Lietuva
Amgen Switzerland AG Vilniaus filialas
Tel.: +370 682 28282
This leaflet was last approved in.
99
Tél: +33 (0)1 40 88 27 00
Detailed information on this medicine is available on the European Medicines Agency web site:
---------------------------------------------------------------------------------------------------------------------------
The following information is intended for medical or healthcare professionals only:
Nplate is a sterile but unpreserved product and is intended for single use only. Nplate should be
reconstituted in accordance with good aseptic practice.
•
Nplate 250 micrograms powder for solution for injection
should be reconstituted with
0.72 ml sterile water for injections, yielding a deliverable volume of 0.5 ml. An additional
overfill is included in each vial to ensure that 250 µg of romiplostim can be delivered.
or
•
Nplate 500 micrograms powder for solution for injection
should be reconstituted with 1.2 ml
sterile water for injections, yielding a deliverable volume of 1 ml. An additional overfill is
included in each vial to ensure that 500 µg of romiplostim can be delivered.
Sodium chloride solutions or bacteriostatic water should not be used when reconstituting the medicinal
product. Water for injections should be injected into the vial. The vial contents may be swirled gently
and inverted during dissolution.
The vial should not be shaken or vigorously agitated.
Generally,
dissolution of Nplate takes less than 2 minutes. Visually inspect the solution for particulate matter and
discolouration before administration. The reconstituted solution should be clear and colourless and
should not be administered if particulate matter and/or discolouration are observed.
From a microbiological point of view, the product should be used immediately. If not used
immediately, in-use storage times and conditions prior to use are the responsibility of the user and
would normally not be longer than 24 hours at 25°C or 24 hours in a refrigerator (2°C – 8°C),
protected from light.
Any unused product or waste material should be disposed of in accordance with local requirements.
1.
Remove the plastic cap from Nplate powder vial and
clean rubber stopper using the provided alcohol swab.
2.
Attach vial adapter to Nplate vial by peeling off
paper
backing from vial adapter, keeping the vial adapter in
its packaging.
Keeping the
vial
on the bench,
push the
vial adapter down onto the centre of the vial until it is
firmly in place.
Note: To prevent contamination of the product, do
not touch the vial adapter spike or Luer lock.
3.
Remove and discard vial adapter packaging.
4.
Attach plunger rod to the pre-filled syringe of water
for injections
by twisting the plunger rod clockwise onto
the syringe plunger, until you feel a slight resistance.
100








5.
Holding the pre-filled syringe of water for injections
with one hand, bend the tip of the white plastic cover
downward with your other hand. This will break the
seal of the white plastic cover. Once the seal is
broken, pull cover off to separate the grey rubber cap
from the clear plastic syringe tip.
6.
Keeping the vial on the bench, attach the pre-filled
syringe of water for injections to vial adapter:
hold the
outer edge of the vial adapter with one hand and twist the
syringe tip clockwise onto the adapter with the other hand
until you feel a slight resistance.
7.
Very slowly and gently expel all water
into powder vial.
Water should flow slowly onto powder. GENTLY swirl
the vial until all of the powder has dissolved and the fluid
in the vial is clear and colourless.
Do not
shake or agitate vial
NOTE:
This may take up to 2
minutes for the powder to
completely dissolve.
Note: From a microbiological point of view, the
product must be used immediately after
reconstitution. If reconstituted product is not used
immediately, the syringe should not be removed from
the vial adapter to maintain microbiological integrity.
Before continuing:
Do
visually inspect the reconstituted solution for particulate matter and/or discoloration. The
reconstituted solution should be clear and colourless and should not be administered if particulate
matter and/or discolouration are observed.
Do
make sure solution is fully dissolved before removing syringe.
8.
Remove the empty pre-filled syringe from the vial
adapter.
9.
Remove 1 ml administration syringe from package.
Attach the 1 ml syringe to
vial adapter of
reconstituted
solution
by twisting the syringe tip onto the vial adapter
until you feel a slight resistance.
10.
Turn assembled syringe-vial unit upside down,
so the
vial of reconstituted product is above the syringe.
Withdraw all
of the medicinal product solution into the
administration
syringe.
101


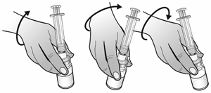











11.
Ensure the correct amount of solution
for the patient
dose is in the administration
syringe by expelling any
excess solution back into the vial.
Note: Remove all air bubbles from syringe to ensure
precise solution amount is in syringe.
12.
Twist off administration syringe from vial adapter.
Attach safety needle
to the filled administration syringe
by twisting needle
clockwise
into syringe Luer lock tip.
13.
Prepare injection site with a new alcohol swab.
Pull
back on the pink safety cover
toward the syringe and
away from the needle.
Remove clear needle shield from prepared needle
by
holding syringe in one hand and carefully pulling shield
straight off with the other hand.
14.
Administer subcutaneous injection
following local
protocols and good aseptic technique.
15.
After injecting, activate the pink safety cover
by
pushing the cover forward using the same hand until you
hear and/or feel it click/lock.
16.
Immediately discard syringe and needle
into an
approved Sharps Container.
102












Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/nplate.html
Copyright © 1995-2021 ITA all rights reserved.