










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Prolia 60 mg solution for injection in a pre-filled syringe
2.
Each pre-filled syringe contains 60 mg of denosumab in 1 ml of solution (60 mg/ml).
Denosumab is a human monoclonal IgG2 antibody produced in a mammalian cell line (CHO) by
recombinant DNA technology.
Excipients known to have a recognised action:
Each ml of solution contains 47 mg sorbitol (E420) (see section 4.4).
For a full list of excipients, see section 6.1.
3.
Solution for injection (injection).
Clear, colourless to slightly yellow solution.
4.
Treatment of osteoporosis in postmenopausal women at increased risk of fractures. Prolia
significantly reduces the risk of vertebral, non vertebral and hip fractures.
Treatment of bone loss associated with hormone ablation in men with prostate cancer at increased risk
of fractures (see section 5.1). In men with prostate cancer receiving hormone ablation, Prolia
significantly reduces the risk of vertebral fractures.
Posology
The recommended dose of Prolia is 60 mg administered as a single subcutaneous injection once every
6 months into the thigh, abdomen or back of arm.
Patients must be adequately supplemented with calcium and vitamin D (see section 4.4).
Patients with renal impairment
No dose adjustment is required in patients with renal impairment (see sections 4.4 and 5.2).
Patients with hepatic impairment
The safety and efficacy of denosumab have not been studied in patients with hepatic impairment (see
section 5.2).
Elderly Patients (age
≥
65)
No dose adjustment is required in elderly patients.
2
Paediatric population
Prolia is not recommended in paediatric patients (age < 18)
as the safety and efficacy of Prolia in these
patients have not been established. Inhibition of RANK/RANK ligand (RANKL) in animal studies has
been coupled to inhibition of bone growth and lack of tooth eruption (see also section 5.3).
Method of administration
Administration should be performed by an individual who has been adequately trained in injection
techniques. For subcutaneous use.
The instructions for use, handling and disposal are given in section 6.6.
-
Hypocalcaemia (see section 4.4).
-
Hypersensitivity to the active substance or to any of the excipients.
Calcium and Vitamin D supplementation
Adequate intake of calcium and vitamin D is important in all patients.
Precautions for use
Hypocalcaemia must be corrected by adequate intake of calcium and vitamin D before initiating
therapy. Patients with severe renal impairment (creatinine clearance < 30 ml/min) or receiving
dialysis are at greater risk of developing hypocalcaemia. Clinical monitoring of calcium levels is
recommended for patients predisposed to hypocalcaemia.
Patients receiving Prolia may develop skin infections (predominantly cellulitis) leading to
hospitalisation (see section 4.8). Patients should be advised to seek prompt medical attention if they
develop signs or symptoms of cellulitis.
Osteonecrosis of the jaw
(
ONJ) has been reported in patients treated with denosumab
or
bisphosphonates, another class of anti-resorptive agents. Most cases have been in cancer patients;
however some have occurred in patients with osteoporosis.
ONJ has been reported rarely in clinical studies in patients receiving denosumab at a dose of 60 mg
every 6 months for osteoporosis.
There have been reports of ONJ in clinical studies in patients with advanced cancer treated with
denosumab at the studied dose of 120 mg administered monthly. Known risk factors for ONJ include
a diagnosis of cancer with bone lesions, concomitant therapies (e.g., chemotherapy, antiangiogenic
biologics, corticosteroids, radiotherapy to head and neck), poor oral hygiene, dental extractions, and
co-morbid disorders (e.g., pre-existing dental disease, anaemia,
coagulopathy, infection) and previous
treatment with bisphosphonates.
A dental examination with appropriate preventive dentistry should be considered prior to treatment
with Prolia in patients with concomitant risk factors. While on treatment, these patients should avoid
invasive dental procedures if possible.
Good oral hygiene practices should be maintained during treatment with Prolia. For patients who
develop ONJ while on Prolia therapy, dental surgery may exacerbate the condition. If ONJ occurs
during treatment with Prolia, use clinical judgment and guide the management plan of each patient
based on individual benefit/risk evaluation.
3
The needle cover of the pre-filled syringe contains dry natural rubber (a derivative of latex), which
may cause allergic reactions.
Warnings for Excipients
Patients with rare hereditary problems of fructose intolerance should not use Prolia.
This medicinal product contains less than 1 mmol sodium (23 mg) per 60 mg i.e. essentially ‘sodium-
free’.
No interaction studies have been performed.
There are no clinical data on the co-administration of denosumab and hormone replacement therapy
(oestrogen), however the potential for a pharmacodynamic interaction is considered to be low.
In postmenopausal women with osteoporosis the pharmacokinetics and pharmacodynamics of
denosumab were not altered by previous alendronate therapy, based on data from a transition study
(alendronate to denosumab).
Pregnancy
There are no adequate data from the use of Prolia in pregnant women. Animal studies do not indicate
direct or indirect harmful effects with respect to reproductive toxicity (see section 5.3). In genetically
engineered mice in which RANKL has been turned off by gene removal (a “knockout mouse”), studies
suggest absence of RANKL (the target of denosumab – see section 5.1) could interfere with the
development of lymph nodes in the foetus and could lead to postnatal impairment of dentition and
bone growth (see section 5.3). Prolia is not recommended for use in pregnant women.
Breast-feeding
It is unknown whether denosumab is excreted in human milk. Knockout mouse studies suggest
absence of RANKL during pregnancy may interfere with maturation of the mammary gland leading to
impaired lactation post-partum (see section 5.3). A decision on whether to abstain from breast-feeding
or to abstain from therapy with Prolia should be made, taking into account the benefit of breast-
feeding to the newborn/infant and the benefit of Prolia therapy to the woman.
Fertility
No data are available on the effect of denosumab on human fertility. Animal studies do not indicate
direct or indirect harmful effects with respect to fertility (see section 5.3).
Prolia has no or negligible influence on the ability to drive and use machines.
Tabulated summary of adverse reactions
The safety of Prolia was evaluated in 10,534 postmenopausal women with osteoporosis (up to 5 years
duration) and breast or prostate cancer patients receiving hormone ablation in phase II and III placebo-
controlled clinical trials.
The following convention has been used for the classification of the adverse reactions reported in
these phase II and III clinical studies (see table 1): very common (≥ 1/10), common (≥ 1/100 to
< 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000) and very rare (< 1/10,000)
based on 1-year event rates. Within each frequency grouping and system organ class, undesirable
effects are presented in order of decreasing seriousness.
4
Table 1 Adverse reactions reported in phase II and phase III placebo-controlled clinical studies
in women with postmenopausal osteoporosis and breast or prostate cancer patients receiving
hormone ablation
MedDRA system organ class
Frequency category
Undesirable effect
Infections and infestations
Common
Common
Uncommon
Uncommon
Uncommon
Urinary tract infection
Upper respiratory tract infection
Diverticulitis
1
Cellulitis
1
Ear infection
Metabolism and nutrition
disorders
Very rare
Hypocalcaemia
1
Nervous system disorders
Common
Sciatica
Eye disorders
Common
Cataracts
1
Gastrointestinal disorders
Common
Constipation
Skin and subcutaneous tissue
disorders
Common
Uncommon
Rash
Eczema
Musculoskeletal and connective
tissue disorders
Common
Pain in extremity
1
See section Description of selected adverse reactions
In a pooled analysis of data from all phase II and phase III placebo controlled studies, Influenza-like
illness was reported with an event rate of 0.006 per subject year for denosumab and 0.003 per subject
year for placebo. Although this imbalance was identified via the pooled analysis, it was not identified
via the stratified analysis which was used to calculate the adverse reactions reported in table 1. There
were no individual studies in which this imbalance was observed.
Description of selected adverse reactions
Hypocalcaemia
In two phase III placebo-controlled clinical trials in postmenopausal women with osteoporosis,
approximately 0.05% (2 out of 4,050) of patients had declines of serum calcium levels (less than
1.88 mmol/l) following Prolia administration. Declines of serum calcium levels (less than
1.88 mmol/l) were not reported in the two phase III placebo-controlled clinical trials in patients
receiving hormone ablation.
Skin infections
In phase III placebo-controlled clinical trials, the overall incidence of skin infections was similar in the
placebo and the Prolia groups in postmenopausal women with osteoporosis (placebo [1.2%, 50 out of
4,041] versus Prolia [1.5%, 59 out of 4,050]) and in breast or prostate cancer patients receiving
hormone ablation (placebo [1.7%, 14 out of 845] versus Prolia [1.4%, 12 out of 860]). Skin infections
leading to hospitalisation were reported in 0.1% (3 out of 4,041) of postmenopausal women with
osteoporosis receiving placebo versus 0.4% (16 out of 4,050) of women receiving Prolia. These cases
were predominantly cellulitis. Skin infections reported as serious adverse reactions were similar in the
placebo (0.6%, 5 out of 845) and the Prolia (0.6%, 5 out of 860) groups in the breast and prostate
cancer studies.
Osteonecrosis of the jaw
In the osteoporosis clinical trial program (8710 patients treated ≥ 1 year), ONJ was reported rarely
with Prolia (see section 4.4).
Cataracts
In a single phase III placebo-controlled clinical trial in patients with prostate cancer receiving
androgen deprivation therapy (ADT) an imbalance in cataract adverse events was observed (4.7%
5












denosumab, 1.2% placebo). No imbalance was observed in postmenopausal women with osteoporosis
or in women undergoing aromatase inhibitor therapy for nonmetastatic breast cancer.
Diverticulitis
In a single phase III placebo-controlled clinical trial in patients with prostate cancer receiving ADT an
imbalance in diverticulitis adverse events was observed (1.2% denosumab, 0% placebo). The
incidence of diverticulitis was comparable between treatment groups in postmenopausal women with
osteoporosis and in women undergoing aromatase inhibitor therapy for nonmetastatic breast cancer.
Other special populations
In clinical studies, patients with severe renal impairment (creatinine clearance < 30 ml/min) or
receiving dialysis were at greater risk of developing hypocalcaemia in the absence of calcium
supplementation. Adequate intake of calcium and vitamin D is important in patients with severe renal
impairment or receiving dialysis (see section 4.4).
There is no experience with overdose in clinical studies. Prolia has been administered in clinical
studies using doses up to 180 mg every 4 weeks (cumulative doses up to 1,080 mg over 6 months),
and no additional adverse reactions were observed.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Drugs for the treatment of bone diseases – Other drugs affecting bone
structure and mineralization, ATC code: M05BX04
Mechanism of action
Denosumab is a human monoclonal antibody (IgG2) that targets and binds with high affinity and
specificity to RANKL, preventing activation of its receptor, RANK, on the surface of osteoclast
precursors and osteoclasts. Prevention of the RANKL/RANK interaction inhibits osteoclast
formation, function and survival, thereby decreasing bone resorption in cortical and trabecular bone.
Pharmacodynamic effects
Prolia treatment rapidly reduced the rate of bone turnover, reaching a nadir for the bone resorption
marker serum type 1 C-telopeptides (CTX) (85% reduction) by 3 days, with reductions maintained
over the dosing interval. At the end of each dosing interval, CTX reductions were partially attenuated
from maximal reduction of ≥ 87% to approximately ≥ 45% (range 45-80%), reflecting the reversibility
of Prolia’s effects on bone remodelling once serum levels diminish. These effects were sustained with
continued treatment. Bone turnover markers generally reached pre-treatment levels within 9 months
after the last dose. Upon re-initiation, reductions in CTX by denosumab were similar to those
observed in patients initiating primary denosumab treatment.
Immunogenicity
In clinical studies, neutralising antibodies have not been observed for Prolia. Using a sensitive
immunoassay < 1% of patients treated with denosumab for up to 5 years tested positive for non
neutralising binding antibodies with no evidence of altered pharmacokinetics, toxicity, or clinical
response.
Treatment of osteoporosis in postmenopausal women
Efficacy and safety of Prolia administered once every 6 months for 3 years were investigated in post-
menopausal women (7,808 women aged 60-91 years, of which 23.6% had prevalent vertebral
fractures) with baseline bone mineral density (BMD) T-scores at the lumbar spine or total hip between
–2.5 and –4.0 and a mean absolute 10-year fracture probability of 18.60% (deciles: 7.9-32.4%) for
6
major osteoporotic fracture and 7.22% (deciles: 1.4-14.9%) for hip fracture. Women with other
diseases or on therapies that may affect bone were excluded from this study. Women received calcium
(at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily.
Effect on vertebral fractures
Prolia significantly reduced the risk of new vertebral fractures at 1, 2 and 3 years (p < 0.0001) (see
table 2).
Table 2 The effect of Prolia on the risk of new vertebral fractures
Proportion of women with fracture (%)
Absolute risk
reduction (%)
(95% CI)
Relative risk
reduction (%)
(95% CI)
Placebo
n = 3,906
Prolia
n = 3,902
0-1 year
2.2
0.9
1.4 (0.8, 1.9)
61 (42, 74)**
0-2 years
5.0
1.4
3.5 (2.7, 4.3)
71 (61,79)**
0-3 years
7.2
2.3
4.8 (3.9, 5.8)
68 (59, 74)*
*p < 0.0001, **p < 0.0001 – exploratory analysis
Effect on hip fractures
Prolia demonstrated a 40% relative reduction (0.5% absolute risk reduction) in the risk of hip fracture
over 3 years (p < 0.05). The incidence of hip fracture was 1.2% in the placebo group compared to
0.7% in the Prolia group at 3 years.
In a post-hoc analysis in women > 75 years, a 62% relative risk reduction was observed with Prolia
(1.4% absolute risk reduction, p < 0.01).
Effect on all clinical fractures
Prolia significantly reduced fractures across all fracture types/groups (see table 3).
Table 3 The effect of Prolia on the risk of clinical fractures over 3 years
Proportion of women with
fracture (%)
+
Absolute risk
reduction (%)
(95% CI)
Relative risk
reduction (%)
(95% CI)
Placebo
n = 3,906
Prolia
n = 3,902
Any clinical fracture
1
10.2 7.2 2.9 (1.6, 4.2) 30 (19, 41)***
Clinical vertebral fracture 2.6 0.8 1.8 (1.2, 2.4) 69 (53, 80)***
Non-vertebral fracture
2
8.0 6.5 1.5 (0.3, 2.7) 20 (5, 33)**
Major non-vertebral fracture
3
6.4 5.2 1.2 (0.1, 2.2) 20 (3, 34)*
Major osteoporotic fracture
4
8.0 5.3 2.7 (1.6, 3.9) 35 (22, 45)***
*p ≤ 0.05; **p = 0.0106
(secondary endpoint included in multiplicity adjustment),
***p ≤ 0.0001
+ Event rates based on Kaplan-Meier estimates at 3 years.
(1) Includes clinical vertebral fractures and non-vertebral fractures.
(2) Excludes those of the vertebrae, skull, facial, mandible, metacarpus, and finger and toe phalanges.
(3) Includes pelvis, distal femur, proximal tibia, ribs, proximal humerus, forearm, and hip.
(4) Includes clinical vertebral, hip, forearm, and humerus fractures, as defined by the WHO.
In women with baseline femoral neck BMD ≤ -2.5, Prolia reduced the risk of non-vertebral fracture
(35% relative risk reduction, 4.1% absolute risk reduction, p < 0.001, exploratory analysis).
The reduction in the incidence of new vertebral fractures, hip fractures and non-vertebral fractures by
Prolia over 3 years were consistent regardless of the 10-year baseline fracture risk.
Effect on bone mineral density
Prolia significantly increased BMD at all clinical sites measured, versus placebo at 1, 2 and 3 years.
Prolia increased BMD by 9.2% at the lumbar spine, 6.0% at the total hip, 4.8% at the femoral neck,
7






















7.9% at the hip trochanter, 3.5% at the distal 1/3 radius and 4.1% at the total body over 3 years (all
p
<
0.0001).
In clinical studies examining the effects of discontinuation of Prolia, BMD returned to approximately
pre-treatment levels and remained above placebo within 18 months of the last dose. These data
indicate that continued treatment with Prolia is required to maintain the effect of the medicinal
product. Re-initiation of Prolia resulted in gains in BMD similar to those when Prolia was first
administered.
Bone histology
Bone histology was evaluated in 62 postmenopausal women with osteoporosis or with low bone mass
who had transitioned from previous alendronate therapy following 1-3 years treatment with Prolia.
Bone biopsy results from both studies showed bone of normal architecture and quality with no
evidence of mineralisation defects, woven bone or marrow fibrosis.
Treatment of bone loss associated with androgen deprivation
Efficacy and safety of Prolia once every 6 months for 3 years were investigated in men with
histologically confirmed non-metastatic prostate cancer receiving ADT (1,468 men aged 48-97 years)
who were at increased risk of fracture (defined as > 70 years, or < 70 years with a BMD T-score at the
lumbar spine, total hip, or femoral neck < -1.0 or a history of an osteoporotic fracture.) All men
received calcium (at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily.
Prolia significantly increased BMD at all clinical sites measured, relative to treatment with placebo at
3 years: 7.9% at the lumbar spine, 5.7% at the total hip, 4.9% at the femoral neck, 6.9% at the hip
trochanter, 6.9% at the distal 1/3 radius and 4.7% at the total body (all p
<
0.0001). In a prospectively
planned exploratory analysis, significant increases in BMD were observed at the lumbar spine, total
hip, femoral neck and the hip trochanter 1 month after the initial dose.
Prolia demonstrated a significant relative risk reduction of new vertebral fractures at 1 year: 85%
(1.6% absolute risk reduction) at 1 year, 69% (2.2% absolute risk reduction) at 2 years and 62% (2.4%
absolute risk reduction) at 3 years (all p < 0.01).
Treatment of bone loss associated with adjuvant aromatase inhibitor therapy
Efficacy and safety of Prolia once every 6 months for 2 years was investigated in women with non-
metastatic breast cancer (252 women aged 35-84 years) and baseline BMD T-scores between
-1.0 to -2.5 at the lumbar spine, total hip or femoral neck. All women received calcium (at least
1,000 mg) and vitamin D (at least 400 IU) supplementation daily.
The primary efficacy variable was percent change in lumbar spine BMD, fracture efficacy was not
evaluated
.
Prolia significantly increased BMD at all clinical sites measured, relative to treatment with
placebo at 2 years: 7.6% at lumbar spine, 4.7% at total hip, 3.6% at femoral neck, 5.9% at hip
trochanter, 6.1% at distal 1/3 radius and 4.2% at total body (all p < 0.0001).
The European Medicines Agency has waived the obligation to submit the results of studies with Prolia
in all subsets of the paediatric population in the treatment of menopausal and other perimenopausal
disorders, and in the treatment of bone loss associated with sex hormone ablative therapy. See 4.2 for
information on paediatric use.
5.2
Pharmacokinetic properties
Following subcutaneous administration of a 1.0 mg/kg dose, which approximates the approved 60 mg
dose, exposure based on AUC was 78% as compared to intravenous administration at the same dose
level. For a 60 mg subcutaneous dose, maximum serum denosumab concentrations (C
max
) of 6 μg/ml
(range 1-17 μg/ml) occurred in 10 days (range 2-28 days). After C
max
, serum levels declined with a
half-life of 26 days (range 6-52 days) over a period of 3 months (range 1.5-4.5 months). Fifty-three
percent (53%) of patients had no measurable amounts of denosumab detected at 6 months post-dose.
In dose ranging studies, denosumab exhibited nonlinear, dose-dependent pharmacokinetics, with lower
8
clearance at higher doses or concentrations, but approximately dose-proportional increases in
exposures for doses of 60 mg and greater.
No accumulation or change in denosumab pharmacokinetics with time was observed upon
subcutaneous multiple-dosing of 60 mg once every 6 months. Denosumab pharmacokinetics was not
affected by the formation of binding antibodies to denosumab and was similar in men and women.
Age (28-87 years)
,
race and disease state (low bone mass or osteoporosis; prostate or breast cancer) do
not appear to significantly affect the pharmacokinetics of denosumab.
A trend was observed between higher body weight and lower exposure based on AUC and C
max
.
However, the trend is not considered clinically important, since pharmacodynamic effects based on
bone turnover markers and BMD increases were consistent across a wide range of body weight.
Denosumab is composed solely of amino acids and carbohydrates as native immunoglobulin and is
unlikely to be eliminated via hepatic metabolic mechanisms. Its metabolism and elimination are
expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides
and individual amino acids.
Special populations
In a study of 55 patients with varying degrees of renal function, including patients on dialysis, the
degree of renal impairment had no effect on the pharmacokinetics of denosumab.
No specific study in patients with hepatic impairment was performed. In general, monoclonal
antibodies are not eliminated via hepatic metabolic mechanisms. The pharmacokinetics of denosumab
is not expected to be affected by hepatic impairment.
The pharmacokinetic profile in paediatric populations has not been assessed.
5.3
Preclinical safety data
In single and repeated dose toxicity studies in cynomolgus monkeys, denosumab doses resulting in
100 to 150 times greater systemic exposure than the recommended human dose had no impact on
cardiovascular physiology, male or female reproduction, or produced specific target organ toxicity.
Standard tests to investigate the genotoxicity potential of denosumab have not been evaluated, since
such tests are not relevant for this molecule. However, due to its character it is unlikely that
denosumab has any potential for genotoxicity.
The carcinogenic potential of denosumab has not been evaluated in long-term animal studies.
At exposures up to 100-fold higher than the human exposure, denosumab showed no evidence of
impaired female fertility and harm to the foetus in cynomolgus monkeys in development toxicity
studies. In preclinical studies conducted in knockout mice lacking RANK or RANKL, impairment of
lymph node formation was observed in the foetus. An absence of lactation due to inhibition of
mammary gland maturation (lobulo-alveolar gland development during pregnancy) was also observed
in knockout mice lacking RANK or RANKL.
In preclinical bone quality studies in monkeys on long-term denosumab treatment, decreases in bone
turnover were associated with improvement in bone strength and normal bone histology. Calcium
levels were transiently decreased and parathyroid hormone levels transiently increased in
ovariectomised monkeys treated with denosumab.
In male mice genetically engineered to express huRANKL (knock-in mice), which were subjected to a
transcortical fracture, denosumab delayed the removal of cartilage and remodelling of the fracture
callus compared to control, but biomechanical strength was not adversely affected.
9
Knockout mice (see section 4.6) lacking RANK or RANKL exhibited decreased body weight, reduced
bone growth and lack of tooth eruption. In neonatal rats, inhibition of RANKL (target of denosumab
therapy) with high doses of a construct of osteoprotegerin bound to Fc (OPG-Fc) was associated with
inhibition of bone growth and tooth eruption. The reversibility of the effects of OPG-Fc has not been
examined. Adolescent primates dosed with denosumab at 27 and 150 times (10 and 50 mg/kg dose)
the clinical exposure had abnormal growth plates. Therefore, treatment with denosumab may impair
bone growth in children with open growth plates and may inhibit eruption of dentition.
6.
6.1
List of excipients
Glacial acetic acid*
Sodium hydroxide (for pH adjustment)*
Sorbitol (E420)
Polysorbate 20
Water for injections
* Acetate buffer is formed by mixing acetic acid with sodium hydroxide
6.2
Incompatibilities
This medicinal product must not be mixed with other medicinal products.
6.3
Shelf life
30 months.
Prolia may be stored at room temperature (up to 25°C) for up to 30 days in the original container.
Once removed from the refrigerator, Prolia must be used within this 30 day period.
6.4
Special precautions for storage
Store in a refrigerator (2°C – 8°C).
Do not freeze.
Keep the pre-filled syringe in the outer carton in order to protect from light.
Do not shake excessively.
6.5
Nature and contents of container
One ml solution in a single use pre-filled syringe made from type I glass with stainless steel 27 gauge
needle, with or without needle guard.
The needle cover of the pre-filled syringe contains dry natural rubber, which is a derivative of latex
(see section 4.4).
Pack size of one, presented in blistered (pre-filled syringe with or without a needle guard) or
unblistered packaging (pre-filled syringe only).
6.6
Special precautions for disposal and other handling
Before administration, the Prolia solution should be inspected
.
Do not inject the solution if it contains
particles, or is cloudy or discoloured. Do not shake excessively. To avoid discomfort at the site of
injection, allow the pre-filled syringe to reach room temperature (up to 25°C) before injecting and
inject slowly. Inject the entire contents of the pre-filled syringe. Dispose of any medicinal product
remaining in the pre-filled syringe.
10
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Amgen Europe B.V.
Minervum 7061
NL-4817 ZK Breda
The Netherlands
8.
9.
Detailed information on this medicinal product is available on the website of the European Medicines
Agency http://www.ema.europa.eu
11
1.
Prolia 60 mg solution for injection
2.
Each vial contains 60 mg of denosumab in 1 ml of solution (60 mg/ml).
Denosumab is a human monoclonal IgG2 antibody produced in a mammalian cell line (CHO) by
recombinant DNA technology.
Excipients known to have a recognised action:
Each ml of solution contains 47 mg sorbitol (E420) (see section 4.4).
For a full list of excipients, see section 6.1.
3.
Solution for injection (injection).
Clear, colourless to slightly yellow solution and may contain trace amounts of translucent to white
proteinaceous particles.
4.
Treatment of osteoporosis in postmenopausal women at increased risk of fractures. Prolia significantly
reduces the risk of vertebral, non vertebral and hip fractures.
Treatment of bone loss associated with hormone ablation in men with prostate cancer at increased risk
of fractures (see section 5.1). In men with prostate cancer receiving hormone ablation, Prolia
significantly reduces the risk of vertebral fractures.
Posology
The recommended dose of Prolia is 60 mg administered as a single subcutaneous injection once every
6 months into the thigh, abdomen or back of arm.
Patients must be adequately supplemented with calcium and vitamin D (see section 4.4).
Patients with renal impairment
No dose adjustment is required in patients with renal impairment (see sections 4.4 and 5.2).
Patients with hepatic impairment
The safety and efficacy of denosumab have not been studied in patients with hepatic impairment (see
section 5.2).
Elderly Patients (age
≥
65)
No dose adjustment is required in elderly patients.
12
Paediatric population
Prolia is not recommended in paediatric patients (age < 18)
as the safety and efficacy of Prolia in these
patients have not been established. Inhibition of RANK/RANK ligand (RANKL) in animal studies
has been coupled to inhibition of bone growth and lack of tooth eruption (see also section 5.3).
Method of administration
Administration should be performed by an individual who has been adequately trained in injection
techniques. For subcutaneous use.
The instructions for use, handling and disposal are given in section 6.6.
-
Hypocalcaemia (see section 4.4).
-
Hypersensitivity to the active substance or to any of the excipients.
Calcium and Vitamin D supplementation
Adequate intake of calcium and vitamin D is important in all patients.
Precautions for use
Hypocalcaemia must be corrected by adequate intake of calcium and vitamin D before initiating
therapy. Patients with severe renal impairment (creatinine clearance < 30 ml/min) or receiving
dialysis are at greater risk of developing hypocalcaemia. Clinical monitoring of calcium levels is
recommended for patients predisposed to hypocalcaemia.
Patients receiving Prolia may develop skin infections (predominantly cellulitis) leading to
hospitalisation (see section 4.8). Patients should be advised to seek prompt medical attention if they
develop signs or symptoms of cellulitis.
Osteonecrosis of the jaw
(
ONJ) has been reported in patients treated with denosumab
or
bisphosphonates, another class of anti-resorptive agents. Most cases have been in cancer patients;
however some have occurred in patients with osteoporosis.
ONJ has been reported rarely in clinical studies in patients receiving denosumab at a dose of 60 mg
every 6 months for osteoporosis.
There have been reports of ONJ in clinical studies in patients with advanced cancer treated with
denosumab at the studied dose of 120 mg administered monthly. Known risk factors for ONJ include
a diagnosis of cancer with bone lesions, concomitant therapies (e.g., chemotherapy, antiangiogenic
biologics, corticosteroids, radiotherapy to head and neck), poor oral hygiene, dental extractions, and
co-morbid disorders (e.g., pre-existing dental disease, anaemia,
coagulopathy, infection) and previous
treatment with bisphosphonates.
A dental examination with appropriate preventive dentistry should be considered prior to treatment
with Prolia in patients with concomitant risk factors. While on treatment, these patients should avoid
invasive dental procedures if possible.
Good oral hygiene practices should be maintained during treatment with Prolia. For patients who
develop ONJ while on Prolia therapy, dental surgery may exacerbate the condition. If ONJ occurs
during treatment with Prolia, use clinical judgment and guide the management plan of each patient
based on individual benefit/risk evaluation.
13
The needle cover of the pre-filled syringe contains dry natural rubber (a derivative of latex), which
may cause allergic reactions.
Warnings for Excipients
Patients with rare hereditary problems of fructose intolerance should not use Prolia.
This medicinal product contains less than 1 mmol sodium (23 mg) per 60 mg i.e. essentially ‘sodium-
free’.
No interaction studies have been performed.
There are no clinical data on the co-administration of denosumab and hormone replacement therapy
(oestrogen), however the potential for a pharmacodynamic interaction is considered to be low.
In postmenopausal women with osteoporosis the pharmacokinetics and pharmacodynamics of
denosumab were not altered by previous alendronate therapy, based on data from a transition study
(alendronate to denosumab).
Pregnancy
There are no adequate data from the use of Prolia in pregnant women. Animal studies do not indicate
direct or indirect harmful effects with respect to reproductive toxicity (see section 5.3). In genetically
engineered mice in which RANKL has been turned off by gene removal (a “knockout mouse”), studies
suggest absence of RANKL (the target of denosumab – see section 5.1) could interfere with the
development of lymph nodes in the foetus and could lead to postnatal impairment of dentition and
bone growth (see section 5.3). Prolia is not recommended for use in pregnant women.
Breast-feeding
It is unknown whether denosumab is excreted in human milk. Knockout mouse studies suggest
absence of RANKL during pregnancy may interfere with maturation of the mammary gland leading to
impaired lactation post-partum (see section 5.3). A decision on whether to abstain from breast-feeding
or to abstain from therapy with Prolia should be made, taking into account the benefit of breast-
feeding to the newborn/infant and the benefit of Prolia therapy to the woman.
Fertility
No data are available on the effect of denosumab on human fertility. Animal studies do not indicate
direct or indirect harmful effects with respect to fertility (see section 5.3).
Prolia has no or negligible influence on the ability to drive and use machines.
Tabulated summary of adverse reactions
The safety of Prolia was evaluated in 10,534 postmenopausal women with osteoporosis (up to 5 years
duration) and breast or prostate cancer patients receiving hormone ablation in phase II and III placebo-
controlled clinical trials.
The following convention has been used for the classification of the adverse reactions reported in
these phase II and III clinical studies (see table 1): very common (≥ 1/10), common (≥ 1/100 to
< 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000) and very rare (< 1/10,000)
based on 1-year event rates. Within each frequency grouping and system organ class, undesirable
effects are presented in order of decreasing seriousness.
14
Table 1 Adverse reactions reported in phase II and phase III placebo-controlled clinical studies
in women with postmenopausal osteoporosis and breast or prostate cancer patients receiving
hormone ablation
MedDRA system organ class
Frequency category
Undesirable effect
Infections and infestations
Common
Common
Uncommon
Uncommon
Uncommon
Urinary tract infection
Upper respiratory tract infection
Diverticulitis
1
Cellulitis
1
Ear infection
Metabolism and nutrition
disorders
Very rare
Hypocalcaemia
1
Nervous system disorders
Common
Sciatica
Eye disorders
Common
Cataracts
1
Gastrointestinal disorders
Common
Constipation
Skin and subcutaneous tissue
disorders
Common
Uncommon
Rash
Eczema
Musculoskeletal and connective
tissue disorders
Common
Pain in extremity
1
See section Description of selected adverse reactions
In a pooled analysis of data from all phase II and phase III placebo controlled studies, Influenza-like
illness was reported with an event rate of 0.006 per subject year for denosumab and 0.003 per subject
year for placebo. Although this imbalance was identified via the pooled analysis, it was not identified
via the stratified analysis which was used to calculate the adverse reactions reported in table 1. There
were no individual studies in which this imbalance was observed.
Description of selected adverse reactions
Hypocalcaemia
In two phase III placebo-controlled clinical trials in postmenopausal women with osteoporosis,
approximately 0.05% (2 out of 4,050) of patients had declines of serum calcium levels (less than
1.88 mmol/l) following Prolia administration. Declines of serum calcium levels (less than
1.88 mmol/l) were not reported in the two phase III placebo-controlled clinical trials in patients
receiving hormone ablation.
Skin infections
In phase III placebo-controlled clinical trials, the overall incidence of skin infections was similar in the
placebo and the Prolia groups in postmenopausal women with osteoporosis (placebo [1.2%, 50 out of
4,041] versus Prolia [1.5%, 59 out of 4,050]) and in breast or prostate cancer patients receiving
hormone ablation (placebo [1.7%, 14 out of 845] versus Prolia [1.4%, 12 out of 860]). Skin infections
leading to hospitalisation were reported in 0.1% (3 out of 4,041) of postmenopausal women with
osteoporosis receiving placebo versus 0.4% (16 out of 4,050) of women receiving Prolia. These cases
were predominantly cellulitis. Skin infections reported as serious adverse reactions were similar in the
placebo (0.6%, 5 out of 845) and the Prolia (0.6%, 5 out of 860) groups in the breast and prostate
cancer studies.
Osteonecrosis of the jaw
In the osteoporosis clinical trial program (8710 patients treated ≥ 1 year), ONJ was reported rarely
with Prolia (see section 4.4).
Cataracts
In a single phase III placebo-controlled clinical trial in patients with prostate cancer receiving
androgen deprivation therapy (ADT) an imbalance in cataract adverse events was observed (4.7%
15












denosumab, 1.2% placebo). No imbalance was observed in postmenopausal women with osteoporosis
or in women undergoing aromatase inhibitor therapy for nonmetastatic breast cancer.
Diverticulitis
In a single phase III placebo-controlled clinical trial in patients with prostate cancer receiving ADT an
imbalance in diverticulitis adverse events was observed (1.2% denosumab, 0% placebo). The
incidence of diverticulitis was comparable between treatment groups in postmenopausal women with
osteoporosis and in women undergoing aromatase inhibitor therapy for nonmetastatic breast cancer.
Other special populations
In clinical studies, patients with severe renal impairment (creatinine clearance < 30 ml/min) or
receiving dialysis were at greater risk of developing hypocalcaemia in the absence of calcium
supplementation. Adequate intake of calcium and vitamin D is important in patients with severe renal
impairment or receiving dialysis (see section 4.4).
There is no experience with overdose in clinical studies. Prolia has been administered in clinical
studies using doses up to 180 mg every 4 weeks (cumulative doses up to 1,080 mg over 6 months),
and no additional adverse reactions were observed.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Drugs for the treatment of bone diseases – Other drugs affecting bone
structure and mineralization, ATC code: M05BX04
Mechanism of action
Denosumab is a human monoclonal antibody (IgG2) that targets and binds with high affinity and
specificity to RANKL, preventing activation of its receptor, RANK, on the surface of osteoclast
precursors and osteoclasts. Prevention of the RANKL/RANK interaction inhibits osteoclast formation,
function and survival, thereby decreasing bone resorption in cortical and trabecular bone.
Pharmacodynamic effects
Prolia treatment rapidly reduced the rate of bone turnover, reaching a nadir for the bone resorption
marker serum type 1 C-telopeptides (CTX) (85% reduction) by 3 days, with reductions maintained
over the dosing interval. At the end of each dosing interval, CTX reductions were partially attenuated
from maximal reduction of ≥ 87% to approximately ≥ 45% (range 45-80%), reflecting the reversibility
of Prolia’s effects on bone remodelling once serum levels diminish. These effects were sustained with
continued treatment. Bone turnover markers generally reached pre-treatment levels within 9 months
after the last dose. Upon re-initiation, reductions in CTX by denosumab were similar to those
observed in patients initiating primary denosumab treatment.
Immunogenicity
In clinical studies, neutralising antibodies have not been observed for Prolia. Using a sensitive
immunoassay < 1% of patients treated with denosumab for up to 5 years tested positive for non
neutralising binding antibodies with no evidence of altered pharmacokinetics, toxicity, or clinical
response.
Treatment of osteoporosis in postmenopausal women
Efficacy and safety of Prolia administered once every 6 months for 3 years were investigated in post-
menopausal women (7,808 women aged 60-91 years, of which 23.6% had prevalent vertebral
fractures) with baseline bone mineral density (BMD) T-scores at the lumbar spine or total hip between
–2.5 and –4.0 and a mean absolute 10-year fracture probability of 18.60% (deciles: 7.9-32.4%) for
major osteoporotic fracture and 7.22% (deciles: 1.4-14.9%) for hip fracture. Women with other
16
diseases or on therapies that may affect bone were excluded from this study. Women received calcium
(at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily.
Effect on vertebral fractures
Prolia significantly reduced the risk of new vertebral fractures at 1, 2 and 3 years (p < 0.0001) (see
table 2).
Table 2 The effect of Prolia on the risk of new vertebral fractures
Proportion of women with fracture (%)
Absolute risk
reduction (%)
(95% CI)
Relative risk
reduction (%)
(95% CI)
Placebo
n = 3,906
Prolia
n = 3,902
0-1 year
2.2
0.9
1.4 (0.8, 1.9)
61 (42, 74)**
0-2 years
5.0
1.4
3.5 (2.7, 4.3)
71 (61,79)**
0-3 years
7.2
2.3
4.8 (3.9, 5.8)
68 (59, 74)*
*p < 0.0001, **p < 0.0001 – exploratory analysis
Effect on hip fractures
Prolia demonstrated a 40% relative reduction (0.5% absolute risk reduction) in the risk of hip fracture
over 3 years (p < 0.05). The incidence of hip fracture was 1.2% in the placebo group compared to
0.7% in the Prolia group at 3 years.
In a post-hoc analysis in women > 75 years, a 62% relative risk reduction was observed with Prolia
(1.4% absolute risk reduction, p < 0.01).
Effect on all clinical fractures
Prolia significantly reduced fractures across all fracture types/groups (see table 3).
Table 3 The effect of Prolia on the risk of clinical fractures over 3 years
Proportion of women with
fracture (%)
+
Absolute risk
reduction (%)
(95% CI)
Relative risk
reduction (%)
(95% CI)
Placebo
n = 3,906
Prolia
n = 3,902
Any clinical fracture
1
10.2 7.2 2.9 (1.6, 4.2) 30 (19, 41)***
Clinical vertebral fracture 2.6 0.8 1.8 (1.2, 2.4) 69 (53, 80)***
Non-vertebral fracture
2
8.0 6.5 1.5 (0.3, 2.7) 20 (5, 33)**
Major non-vertebral fracture
3
6.4 5.2 1.2 (0.1, 2.2) 20 (3, 34)*
Major osteoporotic fracture
4
8.0 5.3 2.7 (1.6, 3.9) 35 (22, 45)***
*p ≤ 0.05; **p = 0.0106
(secondary endpoint included in multiplicity adjustment),
***p ≤ 0.0001
+ Event rates based on Kaplan-Meier estimates at 3 years.
(1) Includes clinical vertebral fractures and non-vertebral fractures.
(2) Excludes those of the vertebrae, skull, facial, mandible, metacarpus, and finger and toe phalanges.
(3) Includes pelvis, distal femur, proximal tibia, ribs, proximal humerus, forearm, and hip.
(4) Includes clinical vertebral, hip, forearm, and humerus fractures, as defined by the WHO.
In women with baseline femoral neck BMD ≤ -2.5, Prolia reduced the risk of non-vertebral fracture
(35% relative risk reduction, 4.1% absolute risk reduction, p < 0.001, exploratory analysis).
The reduction in the incidence of new vertebral fractures, hip fractures and non-vertebral fractures by
Prolia over 3 years were consistent regardless of the 10-year baseline fracture risk.
Effect on bone mineral density
Prolia significantly increased BMD at all clinical sites measured, versus placebo at 1, 2 and 3 years.
Prolia increased BMD by 9.2% at the lumbar spine, 6.0% at the total hip, 4.8% at the femoral neck,
17






















7.9% at the hip trochanter, 3.5% at the distal 1/3 radius and 4.1% at the total body over 3 years (all
p
<
0.0001).
In clinical studies examining the effects of discontinuation of Prolia, BMD returned to approximately
pre-treatment levels and remained above placebo within 18 months of the last dose. These data
indicate that continued treatment with Prolia is required to maintain the effect of the medicinal
product. Re-initiation of Prolia resulted in gains in BMD similar to those when Prolia was first
administered.
Bone histology
Bone histology was evaluated in 62 postmenopausal women with osteoporosis or with low bone mass
who had transitioned from previous alendronate therapy following 1-3 years treatment with Prolia.
Bone biopsy results from both studies showed bone of normal architecture and quality with no
evidence of mineralisation defects, woven bone or marrow fibrosis.
Treatment of bone loss associated with androgen deprivation
Efficacy and safety of Prolia once every 6 months for 3 years were investigated in men with
histologically confirmed non-metastatic prostate cancer receiving ADT (1,468 men aged 48-97 years)
who were at increased risk of fracture (defined as > 70 years, or < 70 years with a BMD T-score at the
lumbar spine, total hip, or femoral neck < -1.0 or a history of an osteoporotic fracture.) All men
received calcium (at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily.
Prolia significantly increased BMD at all clinical sites measured, relative to treatment with placebo at
3 years: 7.9% at the lumbar spine, 5.7% at the total hip, 4.9% at the femoral neck, 6.9% at the hip
trochanter, 6.9% at the distal 1/3 radius and 4.7% at the total body (all p
<
0.0001). In a prospectively
planned exploratory analysis, significant increases in BMD were observed at the lumbar spine, total
hip, femoral neck and the hip trochanter 1 month after the initial dose.
Prolia demonstrated a significant relative risk reduction of new vertebral fractures at 1 year: 85%
(1.6% absolute risk reduction) at 1 year, 69% (2.2% absolute risk reduction) at 2 years and 62% (2.4%
absolute risk reduction) at 3 years (all p < 0.01).
Treatment of bone loss associated with adjuvant aromatase inhibitor therapy
Efficacy and safety of Prolia once every 6 months for 2 years was investigated in women with non-
metastatic breast cancer (252 women aged 35-84 years) and baseline BMD T-scores between
-1.0 to -2.5 at the lumbar spine, total hip or femoral neck. All women received calcium (at least
1,000 mg) and vitamin D (at least 400 IU) supplementation daily.
The primary efficacy variable was percent change in lumbar spine BMD, fracture efficacy was not
evaluated
.
Prolia significantly increased BMD at all clinical sites measured, relative to treatment with
placebo at 2 years: 7.6% at lumbar spine, 4.7% at total hip, 3.6% at femoral neck, 5.9% at hip
trochanter, 6.1% at distal 1/3 radius and 4.2% at total body (all p < 0.0001).
The European Medicines Agency has waived the obligation to submit the results of studies with Prolia
in all subsets of the paediatric population in the treatment of menopausal and other perimenopausal
disorders, and in the treatment of bone loss associated with sex hormone ablative therapy. See 4.2 for
information on paediatric use.
5.2
Pharmacokinetic properties
Following subcutaneous administration of a 1.0 mg/kg dose, which approximates the approved 60 mg
dose, exposure based on AUC was 78% as compared to intravenous administration at the same dose
level. For a 60 mg subcutaneous dose, maximum serum denosumab concentrations (C
max
) of 6 μg/ml
(range 1-17 μg/ml) occurred in 10 days (range 2-28 days). After C
max
, serum levels declined with a
half-life of 26 days (range 6-52 days) over a period of 3 months (range 1.5-4.5 months). Fifty-three
percent (53%) of patients had no measurable amounts of denosumab detected at 6 months post-dose.
In dose ranging studies, denosumab exhibited nonlinear, dose-dependent pharmacokinetics, with lower
18
clearance at higher doses or concentrations, but approximately dose-proportional increases in
exposures for doses of 60 mg and greater.
No accumulation or change in denosumab pharmacokinetics with time was observed upon
subcutaneous multiple-dosing of 60 mg once every 6 months. Denosumab pharmacokinetics was not
affected by the formation of binding antibodies to denosumab and was similar in men and women.
Age (28-87 years)
,
race and disease state (low bone mass or osteoporosis; prostate or breast cancer) do
not appear to significantly affect the pharmacokinetics of denosumab.
A trend was observed between higher body weight and lower exposure based on AUC and C
max
.
However, the trend is not considered clinically important, since pharmacodynamic effects based on
bone turnover markers and BMD increases were consistent across a wide range of body weight.
Denosumab is composed solely of amino acids and carbohydrates as native immunoglobulin and is
unlikely to be eliminated via hepatic metabolic mechanisms. Its metabolism and elimination are
expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides
and individual amino acids.
Special populations
In a study of 55 patients with varying degrees of renal function, including patients on dialysis, the
degree of renal impairment had no effect on the pharmacokinetics of denosumab.
No specific study in patients with hepatic impairment was performed. In general, monoclonal
antibodies are not eliminated via hepatic metabolic mechanisms. The pharmacokinetics of denosumab
is not expected to be affected by hepatic impairment.
The pharmacokinetic profile in paediatric populations has not been assessed.
5.3
Preclinical safety data
In single and repeated dose toxicity studies in cynomolgus monkeys, denosumab doses resulting in
100 to 150 times greater systemic exposure than the recommended human dose had no impact on
cardiovascular physiology, male or female reproduction, or produced specific target organ toxicity.
Standard tests to investigate the genotoxicity potential of denosumab have not been evaluated, since
such tests are not relevant for this molecule. However, due to its character it is unlikely that
denosumab has any potential for genotoxicity.
The carcinogenic potential of denosumab has not been evaluated in long-term animal studies.
At exposures up to 100-fold higher than the human exposure, denosumab showed no evidence of
impaired female fertility and harm to the foetus in cynomolgus monkeys in development toxicity
studies. In preclinical studies conducted in knockout mice lacking RANK or RANKL, impairment of
lymph node formation was observed in the foetus. An absence of lactation due to inhibition of
mammary gland maturation (lobulo-alveolar gland development during pregnancy) was also observed
in knockout mice lacking RANK or RANKL.
In preclinical bone quality studies in monkeys on long-term denosumab treatment, decreases in bone
turnover were associated with improvement in bone strength and normal bone histology. Calcium
levels were transiently decreased and parathyroid hormone levels transiently increased in
ovariectomised monkeys treated with denosumab.
In male mice genetically engineered to express huRANKL (knock-in mice), which were subjected to a
transcortical fracture, denosumab delayed the removal of cartilage and remodelling of the fracture
callus compared to control, but biomechanical strength was not adversely affected.
19
Knockout mice (see section 4.6) lacking RANK or RANKL exhibited decreased body weight, reduced
bone growth and lack of tooth eruption. In neonatal rats, inhibition of RANKL (target of denosumab
therapy) with high doses of a construct of osteoprotegerin bound to Fc (OPG-Fc) was associated with
inhibition of bone growth and tooth eruption. The reversibility of the effects of OPG-Fc has not been
examined. Adolescent primates dosed with denosumab at 27 and 150 times (10 and 50 mg/kg dose)
the clinical exposure had abnormal growth plates. Therefore, treatment with denosumab may impair
bone growth in children with open growth plates and may inhibit eruption of dentition.
6.
6.1 List of excipients
Glacial acetic acid*
Sodium hydroxide (for pH adjustment)*
Sorbitol (E420)
Water for injections
* Acetate buffer is formed by mixing acetic acid with sodium hydroxide
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products.
6.3 Shelf life
30 months.
Prolia may be stored at room temperature (up to 25°C) for up to 30 days in the original container.
Once removed from the refrigerator, Prolia must be used within this 30 day period.
6.4
Special precautions for storage
Store in a refrigerator (2°C – 8°C).
Do not freeze.
Keep the vial in the outer carton in order to protect from light.
Do not shake excessively.
6.5 Nature and contents of container
One ml solution in a single use vial made from type I glass with fluoropolymer coated elastomeric
stopper and aluminium seal with flip-off cap.
Pack size of one.
6.6 Special precautions for disposal and other handling
Before administration, the Prolia solution should be inspected. The solution may contain trace
amounts of translucent to white proteinaceous particles. Do not inject the solution if it is cloudy or
discoloured. Do not shake excessively. To avoid discomfort at the site of injection, allow the vial to
reach room temperature (up to 25°C) before injecting and inject slowly. Inject the entire contents of
the vial. Dispose of any medicinal product remaining in the vial.
A 27 gauge needle is recommended for the administration of denosumab. Do not re-enter the vial.
Any unused product or waste material should be disposed of in accordance with local requirements.
20
7.
Amgen Europe B.V.
Minervum 7061
NL-4817 ZK Breda
The Netherlands
8.
9.
Detailed information on this medicinal product is available on the website of the European Medicines
Agency
http://www.ema.europa.eu
21
ANNEX II
A.
MANUFACTURER OF THE BIOLOGICAL ACTIVE
SUBSTANCE AND MANUFACTURING AUTHORISATION
HOLDER RESPONSIBLE FOR BATCH RELEASE
B.
CONDITIONS OF THE MARKETING AUTHORISATION
22
A. MANUFACTURER OF THE BIOLOGICAL ACTIVE SUBSTANCE AND
MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturer of the biological active substance
Boehringer Ingelheim Pharma GmbH & Co. KG
Birkendorfer Strasse 65
D-88397 Biberach an der Riss
Germany
Amgen Inc.
4000 Nelson Road, Longmont, CO 80503
United States
Amgen Inc.
5550 Airport Boulevard, Boulder, CO 80301
United States
Amgen Inc.
One Amgen Center Drive,
Thousand Oaks, CA 91320
United States
Amgen Manufacturing Limited
PO Box 4060, Road 31 km 24.6,
Juncos, PR 00777-4060
Puerto Rico
Name and address of the manufacturer responsible for batch release
Amgen Europe B.V.
Minervum 7061
NL-4817 ZK Breda
The Netherlands
B. CONDITIONS OF THE MARKETING AUTHORISATION
•
Medicinal product subject to medical prescription.
•
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND
EFFECTIVE USE OF THE MEDICINAL PRODUCT
Not applicable
•
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, as described in version 3.3 presented in
Module 1.8.1. of the Marketing Authorisation Application, is in place and functioning before and
whilst the product is on the market.
23
Risk Management plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in
the Pharmacovigilance Plan, as agreed in version 1.3 from 03 December 2009 of the Risk
Management Plan (RMP) presented in Module 1.8.2. of the Marketing Authorisation Application and
any subsequent updates of the RMP agreed by the CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, the
updated RMP should be submitted at the same time as the next Periodic Safety Update Report
(PSUR).
In addition, an updated RMP should be submitted:
•
When new information is received that may impact on the current Safety Specification,
Pharmacovigilance Plan or risk minimisation activities
•
Within 60 days of an important (pharmacovigilance or risk minimisation) milestone being
reached
•
At the request of the European Medicines Agency.
24
ANNEX III
LABELLING AND PACKAGE LEAFLET
25
A. LABELLING
26
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
PRE-FILLED SYRINGE CARTON
1.
Prolia 60 mg solution for injection in a pre-filled syringe
denosumab
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
1 ml pre-filled syringe containing 60 mg of denosumab (60 mg/ml).
3.
LIST OF EXCIPIENTS
Glacial acetic acid, sodium hydroxide, sorbitol (E420), polysorbate 20 and water for injections.
4.
Solution for injection
One pre-filled syringe with automatic needle guard.
One pre-filled syringe.
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Subcutaneous use.
Read the package leaflet before use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
Do not shake excessively.
8.
EXPIRY DATE
EXP
27























9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator.
Do not freeze.
Store in the original carton in order to protect from light.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Amgen Europe B.V.
Minervum 7061,
NL-4817 ZK Breda,
The Netherlands
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Prolia
28



















MINIMUM PARTICULARS TO APPEAR ON BLISTERS OR STRIPS
BLISTERED PRE-FILLED SYRINGE
1.
Prolia 60 mg injection
denosumab
2.
Amgen Europe B.V.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
SC
29

















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
PRE-FILLED SYRINGE LABEL (UNBLISTERED)
1.
Prolia 60 mg injection
denosumab
SC
2.
METHOD OF ADMINISTRATION
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
1 ml
6.
OTHER
30

















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
PRE-FILLED SYRINGE LABEL (BLISTERED)
1.
Prolia 60 mg
denosumab
SC
2.
METHOD OF ADMINISTRATION
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
1 ml
6.
OTHER
31

















PARTICULARS TO APPEAR ON THE OUTER PACKAGING
VIAL CARTON
1.
Prolia 60 mg solution for injection
denosumab
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
1 ml vial containing 60 mg of denosumab (60 mg/ml).
3.
LIST OF EXCIPIENTS
Glacial acetic acid, sodium hydroxide, sorbitol (E420) and water for injections.
4.
Solution for injection
One vial.
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Subcutaneous use.
Read the package leaflet before use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
Do not shake excessively.
8.
EXPIRY DATE
EXP
32



















9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator.
Do not freeze.
Store in the original carton in order to protect from light.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Amgen Europe B.V.
Minervum 7061,
NL-4817 ZK Breda,
The Netherlands
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Prolia
33



















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
VIAL LABEL
1.
Prolia 60 mg injection
denosumab
SC
2.
METHOD OF ADMINISTRATION
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
1 ml
6.
OTHER
34

















REMINDER STICKERS TEXT (included in pack)
Next injection
Prolia 60 mg injection
denosumab
SC
Every 6 months
Amgen Europe B.V.
<…./…./….>
35



B. PACKAGE LEAFLET
36
PACKAGE LEAFLET: INFORMATION FOR THE USER
Prolia 60 mg solution for injection in a pre-filled syringe
denosumab
Read all of this leaflet carefully before you start using this medicine.
-
Keep this leaflet. You may need to read it again.
-
If you have any further questions, ask your doctor or pharmacist.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
-
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor or pharmacist.
In this leaflet
:
1. What Prolia is and what it is used for
2. Before you use Prolia
3. How to use Prolia
4. Possible side effects
5.
How to store Prolia
6.
Further information
1.
WHAT PROLIA IS AND WHAT IT IS USED FOR
What Prolia is and how it works
Prolia contains denosumab, a protein (monoclonal antibody) that interferes with the action of another
protein, in order to treat bone loss and osteoporosis. Treatment with Prolia makes bone stronger and
less likely to break.
Bone is a living tissue and is renewed all the time. Oestrogen helps keep bones healthy. After the
menopause, oestrogen level drops which may cause bones to become thin and fragile. This can
eventually lead to a condition called osteoporosis. Many women with osteoporosis have no
symptoms, but they are still at risk of breaking bones, especially in the spine, hips and wrists.
Surgery or medicines that stop the production of oestrogen or testosterone used to treat patients with
breast or prostate cancer can also lead to bone loss. The bones become weaker and break more easily.
What Prolia is used for
Prolia is used to treat:
•
osteoporosis in women after the menopause (postmenopausal), reducing the risk of spinal, non-
spinal and hip fractures.
•
bone loss that results from a reduction in hormone (testosterone) level caused by surgery or
treatment with medicines in patients with prostate cancer.
2.
BEFORE YOU USE PROLIA
Do not use Prolia
•
if you have low calcium levels in the blood (hypocalcaemia).
•
if you are allergic (hypersensitive) to denosumab or any of the other ingredients of Prolia.
37
Take special care with Prolia
Please tell your doctor immediately if you develop a swollen, red area of skin, most commonly in the
lower leg, that feels hot and tender (cellulitis), and possibly with symptoms of fever while being on
treatment with Prolia.
Please tell your doctor if you have an allergy to latex (the needle cover on the pre-filled syringe
contains a derivative of latex).
Tell your doctor if you have or have ever had severe kidney problems, kidney failure or have needed
dialysis.
You should also take calcium and vitamin D supplements while being on treatment with Prolia. Your
doctor will discuss this with you.
A dental examination should be considered before you start treatment with Prolia if you have cancer,
are undergoing chemotherapy or radiotherapy, are taking steroids, do not receive routine dental care or
have gum disease.
If you are under dental treatment or will undergo dental surgery, tell your dentist that you are being
treated with Prolia.
It is important to maintain good oral hygiene when being on treatment with Prolia.
Prolia is not recommended for anyone under 18 years of age. The use of Prolia in children and
adolescents has not been studied.
Using other medicines
Please tell your doctor or pharmacist if you are taking or have recently taken any other medicines,
including medicines obtained without a prescription.
Pregnancy and breast-feeding
Prolia has not been tested in pregnant women. It is important to tell your doctor if you are pregnant;
think you may be pregnant; or plan to get pregnant. Prolia is not recommended for use if you are
pregnant.
It is not known whether Prolia is excreted in breast milk. It is important to tell your doctor if you are
breast-feeding or plan to do so. Your doctor will then help you decide whether to stop breast-feeding,
or whether to stop taking Prolia, considering the benefit of breast-feeding to the baby and the benefit
of Prolia to the mother.
Ask your doctor or pharmacist for advice before taking any medicine.
Driving and using machines
Prolia has no or negligible influence on the ability to drive and use machines
Important information about some of the ingredients of Prolia
If you have an intolerance to some sugars
If you have been told by your doctor that you have an intolerance to some sugars (sorbitol E420),
contact your doctor before taking this medicinal product.
38
If you are on a controlled sodium diet
This medicinal product contains less than 1 mmol sodium (23 mg) per 60 mg, i.e. essentially ‘sodium-
free’.
3.
HOW TO USE PROLIA
The usual dose is one pre-filled syringe of 60 mg administered once every 6 months, as a single
injection under the skin (subcutaneous). The best places to inject are the top of
your thighs and the abdomen. Your carer can also use the outer area of your
upper arm. Each pack of Prolia contains a reminder card with stickers that can
be removed from the carton. Use the peel-off stickers to mark the next injection
date on your personal calendar and/or the reminder card to keep a record of the
next injection date.
You should also take calcium and vitamin D supplements while being on treatment with Prolia. Your
doctor will discuss this with you.
Your doctor may decide that it is best for you or a carer to inject Prolia. Your doctor or healthcare
provider will show you or your carer how to use Prolia. For instructions on how to inject Prolia,
please read the section at the end of this leaflet.
If you forget to use Prolia
If a dose of Prolia is missed, the injection should be administered as soon as possible. Thereafter,
injections should be scheduled every 6 months from the date of the last injection.
If you stop using Prolia
To get the most benefit from your treatment, it is important to use Prolia for as long as your doctor
prescribes it for you. Please talk to your doctor before you consider stopping the treatment.
4.
POSSIBLE SIDE EFFECTS
Uncommonly, patients receiving Prolia may develop skin infections (predominantly cellulitis).
Please
tell your doctor immediately
if you develop any of these symptoms while being on treatment with
Prolia: swollen, red area of skin, most commonly in the lower leg, that feels hot and tender, and
possibly with symptoms of fever.
Like all medicines, Prolia can cause side effects, although not everybody gets them.
The frequency of possible side effects listed below is defined using the following convention:
Very common (affects more than 1 user in 10)
Common (affects 1 to 10 users in 100)
Uncommon (affects 1 to 10 users in 1,000)
Rare (affects 1 to 10 users in 10,000)
Very rare (affects less than 1 user in 10,000)
Not known (frequency cannot be estimated from the available data).
39

Common side effects
:
•
painful urination, frequent urination, blood in the urine, inability to hold your urine,
•
upper respiratory tract infection,
•
pain, tingling or numbness that moves down your leg (sciatica),
•
cloudy area in the lens of the eye (cataracts),
•
constipation,
•
rash,
•
arm or leg pain (pain in extremity).
Uncommon side effects
:
•
swollen, red area of skin, most commonly in the lower leg, that feels hot and tender (cellulitis),
and possibly with symptoms of fever,
•
fever, vomiting and abdominal pain and discomfort (diverticulitis),
•
ear infection,
•
skin condition with itching, redness and/or dryness (eczema).
Rare side effects:
•
Persistent pain and/or non-healing sores of the mouth or jaw.
Very rare side effects
:
•
low calcium levels in the blood (hypocalcaemia).
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet, please
tell your doctor or pharmacist.
5.
HOW TO STORE PROLIA
Keep out of the reach and sight of children.
Do not use Prolia after the expiry date which is stated on the label and carton after EXP. The expiry
date refers to the last day of that month.
Store in a refrigerator (2°C – 8°C).
Do not freeze.
Store in the original carton in order to protect from light.
Do not shake excessively.
Your pre-filled syringe may be left outside the refrigerator to reach room temperature (up to 25°C)
before injection. This will make the injection more comfortable. Once your syringe has been left to
reach room temperature (up to 25°C), it must be used within 30 days.
Medicines should not be disposed of via wastewater or household waste. Ask your pharmacist how to
dispose of medicines no longer required. These measures will help to protect the environment.
6.
FURTHER INFORMATION
What Prolia contains
-
The active substance is denosumab. Each 1 ml pre-filled syringe contains 60 mg of denosumab
(60 mg/ml).
40
-
The other ingredients are glacial acetic acid, sodium hydroxide, sorbitol (E420), polysorbate 20
and water for injections.
What Prolia looks like and contents of the pack
Prolia is a clear, colourless to slightly yellow solution for injection provided in a ready to use pre-
filled syringe.
Each pack contains one pre-filled syringe with a needle guard.
Each pack contains one pre-filled syringe.
Marketing Authorisation Holder and Manufacturer
Amgen Europe B.V.
Minervum 7061
NL-4817 ZK Breda
The Netherlands
For any information about this medicine, please contact the local representative of the Marketing
Authorisation Holder.
België/Belgique/Belgien
s.a. Amgen n.v.
Tel/Tél: +32 (0)2 7752711
Luxembourg/Luxemburg
s.a. Amgen
Belgique/Belgien
Tel/Tél: +32 (0)2 7752711
България
Амджен България ЕООД
Тел: +359 (0) 2 805 7020
Magyarország
Amgen Kft.
Tel. : +36 1 35 44 700
Česká republika
Amgen s.r.o
Tel : +420 2 21 773 500
Malta
Amgen B.V.
The Netherlands
Tel: +31 (0) 76 5732500
Danmark
Amgen filial af Amgen AB, Sverige
Tlf: +45 39617500
Nederland
Amgen B.V.
Tel: +31 (0) 76 5732500
Deutschland
AMGEN GmbH
Tel: +49 (0)89 1490960
Norge
Amgen AB
Tel: +47 23308000
Eesti
Amgen Switzerland AG Eesti filiaal
Tel: +372 5125 501
Österreich
Amgen GmbH
Tel: +43 (0) 1 50 217
Ελλάδα
Amgen Ελλάς Φαρμακευτικά ΕΠΕ.
Τηλ.: +30 210 3447000
Polska
Amgen Sp. z o.o.
Tel.: +48 22 581 3000
España
Amgen S.A.
Tel: +34 93 600 19 00
Portugal
AMGEN Biofarmacêutica, Lda.
Tel: +351 21 4220550
41
France
Amgen S.A.S
Tél: +33 (0)1 40 88 27 00
România
Mediplus Exim SRL
Tel.: +4021 301 74 74
Ireland
Amgen Limited
United Kingdom
Tel:
+44 (0)1223 420305
Slovenija
AMGEN zdravila d.o.o.
Tel: +386 1 585 1767
Ísland
Vistor hf.
Sími: +354 535 7000
Slovenská republika
Amgen Switzerland AG Slovakia
Tel : +421 33 321 13 22
Italia
Amgen Dompé S.p.A.
Tel: +39 02 6241121
Suomi/Finland
Amgen AB, sivuliike Suomessa/Amgen AB, filial
i Finland
Puh/Tel: +358 (0)9 54900500
Kύπρος
Papaellinas & Co Ltd
Τηλ.: + 357 22741 741
Sverige
Amgen AB
Tel: +46 (0)8 6951100
Latvija
Amgen Switzerland AG Rīgas filiāle
Tel : +371 29284 807
United Kingdom
Amgen Limited
Tel: +44 (0)1223 420305
Lietuva
Amgen Switzerland AG Vilniaus filialas
Tel. +370 6983 6600
This leaflet was last approved in
{MM/YYYY}.
Detailed information on this medicine is available on the European Medicines Agency web site:
---------------------------------------------------------------------------------------------------------------------------
INSTRUCTIONS FOR INJECTING WITH THE PROLIA PRE-FILLED SYRINGE WITH AN
AUTOMATIC NEEDLE GUARD
This section contains information on
how to use the Prolia pre-filled syringe.
It is important that
you or your carer do not give the injection unless training from your doctor or healthcare
provider has been received.
Always wash your hands before every injection. If you have questions
about how to inject, please ask your doctor or healthcare provider for assistance.
Window
Plunger
Needle guard
Needle cover
42
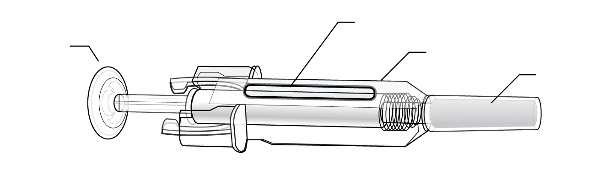

Before you begin
Read all instructions thoroughly before using the pre-filled syringe.
To reduce the risk of accidental needle sticks to users, each pre-filled syringe comes with a needle
guard that is automatically activated to cover the needle after complete delivery of the pre-filled
syringe content.
DO NOT
attempt to activate the needle guard prior to injection.
DO NOT
use the pre-filled syringe if the needle cover has been removed, or the needle guard has been
activated (covering the needle).
How do you use the Prolia pre-filled syringe?
Your doctor has prescribed a Prolia pre-filled syringe for injection into the tissue just under the skin
(subcutaneous). You must inject the entire content (1 ml) of the Prolia pre-filled syringe and it should
be injected once every 6 months as instructed by your doctor or healthcare provider.
Equipment:
To give an injection, you will need:
1.
A new Prolia pre-filled syringe; and
2.
Alcohol wipes or similar.
What to do before you give a subcutaneous injection of Prolia
1.
Remove the pre-filled syringe from the refrigerator.
DO NOT
pick up the pre-filled syringe by the plunger or needle cover. This could damage the
device.
2.
The pre-filled syringe may be left outside the refrigerator to reach room temperature. This will
make the injection more comfortable.
DO NOT
warm it in any other way, for example, in a microwave or in hot water.
DO NOT
leave the syringe exposed to direct sunlight.
3.
DO NOT
shake the pre-filled syringe excessively.
4.
DO NOT
remove the needle cover from the pre-filled syringe until you are ready to inject.
5.
Check the expiry date on the pre-filled syringe label (EXP:).
DO NOT
use it if the date has passed the last day of the month shown.
6.
Check the appearance of Prolia.
It must be a clear, colourless to slightly yellow solution.
The solution should not be injected if it contains particles or if it is cloudy or discoloured.
7.
Find a comfortable, well-lit, clean surface and put all the equipment within reach.
8.
Wash your hands thoroughly.
43
Where should you give the injection?
The best places to inject are the top of your thighs and
the abdomen.
Your carer can also use the outer area of your upper
arms.
How do you give the injection?
1.
Disinfect the skin by using an alcohol wipe.
2.
To avoid bending the needle, gently pull the
cover from the needle
straight off
without
twisting, as shown.
DO NOT
touch the needle or push the plunger.
3.
You may notice a small bubble in the pre-filled
syringe. You do not have to remove the air
bubble before injecting. Injecting the solution
with the air bubble is harmless.
4.
Pinch (without squeezing) the skin between your
thumb and forefinger. Put the needle fully into
the skin as shown by your doctor or healthcare
provider.
5.
Push the plunger with a
slow
constant pressure,
always keeping the skin pinched. Push the
plunger all the way down as far as it will go to
inject
all the solution
.
The needle guard will not activate unless you
empty the pre-filled syringe.
44



















6.
While the plunger is still pressed all the way
down, remove the needle and let go of the skin.
Release the plunger and allow the syringe to
move up until the entire needle is covered by the
needle guard.
7. If the needle guard is not activated, an incomplete
injection may have occurred.
Call your doctor or healthcare provider if you
think you have not received the full dose.
DO NOT
put the needle cover back on used
syringes.
8.
If you notice a spot of blood, you may gently dab
it away with a cotton ball or tissue. Do not rub
the injection site. If needed, you may cover the
injection site with a plaster.
9.
Only use each pre-filled syringe for one injection.
DO NOT
use any Prolia that is left in the syringe.
Remember:
If you have any problems, please ask your doctor or healthcare provider for help and
advice.
Disposing of used syringes
•
DO NOT
put the needle cover back on used syringes.
45









•
Keep used syringes out of the reach and sight of children.
•
The used syringe should be disposed of in accordance with local requirements. Ask your
pharmacist how to dispose of medicines no longer required. These measures will help to protect
the environment.
46
INSTRUCTIONS FOR INJECTING WITH THE PROLIA PRE-FILLED SYRINGE
This section contains information on
how to use the Prolia pre-filled syringe.
It is important that
you or your carer do not give the injection unless training from your doctor or healthcare
provider
has been received.
Always wash your hands before every injection. If you have questions
about how to inject, please ask your doctor or healthcare provider for assistance.
Before you begin
Read all instructions thoroughly before using the pre-filled syringe.
DO NOT
use the pre-filled syringe if the needle cover has been removed.
How do you use the Prolia pre-filled syringe?
Your doctor has prescribed a Prolia pre-filled syringe for injection into the tissue just under the skin
(subcutaneous). You must inject the entire content (1 ml) of the Prolia pre-filled syringe and it should
be injected once every 6 months as instructed by your doctor.
Equipment:
To give an injection, you will need:
1.
A new Prolia pre-filled syringe; and
2.
Alcohol wipes or similar.
What to do before you give a subcutaneous injection of Prolia
1.
Remove the pre-filled syringe from the refrigerator.
DO NOT
pick up the pre-filled syringe by the plunger or needle cover. This could damage the
device.
2.
The pre-filled syringe may be left outside the refrigerator to reach room temperature. This will
make the injection more comfortable.
DO NOT
warm it in any other way, for example, in a microwave or in hot water.
DO NOT
leave the syringe exposed to direct sunlight.
3.
DO NOT
shake the pre-filled syringe excessively.
4.
DO NOT
remove the needle cover from the pre-filled syringe until you are ready to inject.
5.
Check the expiry date on the pre-filled syringe label (EXP:).
DO NOT
use it if the date has passed the last day of the month shown.
6.
Check the appearance of Prolia. It must be a clear, colourless to slightly yellow solution. The
solution should not be injected if it contains particles or if it is discoloured or cloudy.
7.
Find a comfortable, well-lit, clean surface and put all the equipment within reach.
8.
Wash your hands thoroughly.
47

Where should you give the injection?
The best places to inject are the top of your thighs and the
abdomen.
Your carer can also use the outer area of your upper arms.
How do you give the injection?
1.
Disinfect the skin by using an alcohol wipe.
2.
To avoid bending the needle, gently pull the
cover from the needle straight off without
twisting, as shown in pictures 1 and 2.
DO NOT
touch the needle or push the plunger.
3.
You may notice a small air bubble in the pre-
filled syringe. You do not have to remove the
air bubble before injecting. Injecting the
solution with the air bubble is harmless.
4.
Pinch (without squeezing) the skin between
your thumb and forefinger. Put the needle fully
into the skin as shown by your doctor or
healthcare provider.
5.
Push the plunger with a
slow
constant pressure, always keeping the skin pinched. Push the
plunger all the way down as far as it will go to inject
all the solution.
6.
Remove the needle and let go of the skin.
7.
If you notice a spot of blood you may gently dab it away with a cotton ball or tissue. Do not rub
the injection site.
If needed, you may cover the injection site with a plaster.
8.
Only use each pre-filled syringe for one injection.
DO NOT
use any Prolia that is left in the
syringe.
Remember:
if you have any problems, please ask your doctor or healthcare provider for help and
advice.
Disposing of used syringes
•
DO NOT
put the cover back on used needles.
•
Keep used syringes out of the reach and sight of children.
•
The used syringe should be disposed of in accordance with local requirements. Ask your
pharmacist how to dispose of medicines no longer required. These measures will help to protect
the environment.
48



PACKAGE LEAFLET: INFORMATION FOR THE USER
Prolia 60 mg solution for injection
denosumab
Read all of this leaflet carefully before you start using this medicine.
-
Keep this leaflet. You may need to read it again.
-
If you have any further questions, ask your doctor or pharmacist.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
-
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor or pharmacist.
In this leaflet
:
1.
What Prolia is and what it is used for
3.
How to use Prolia
4.
Possible side effects
5.
How to store Prolia
6.
Further information
1.
WHAT PROLIA IS AND WHAT IT IS USED FOR
What Prolia is and how it works
Prolia contains denosumab, a protein (monoclonal antibody) that interferes with the action of another
protein, in order to treat bone loss and osteoporosis. Treatment with Prolia makes bone stronger and
less likely to break.
Bone is a living tissue and is renewed all the time. Oestrogen helps keep bones healthy. After the
menopause, oestrogen level drops which may cause bones to become thin and fragile. This can
eventually lead to a condition called osteoporosis. Many women with osteoporosis have no
symptoms, but they are still at risk of breaking bones, especially in the spine, hips and wrists.
Surgery or medicines that stop the production of oestrogen or testosterone used to treat patients with
breast or prostate cancer can also lead to bone loss. The bones become weaker and break more easily.
What Prolia is used for?
Prolia is used to treat:
•
osteoporosis in women after the menopause (postmenopausal), reducing the risk of spinal, non-
spinal and hip fractures.
•
bone loss that results from a reduction in hormone (testosterone) level caused by surgery or
treatment with medicines in patients with prostate cancer.
2.
BEFORE YOU USE PROLIA
Do not use Prolia
•
if you have low calcium levels in the blood (hypocalcaemia).
•
if you are allergic (hypersensitive) to denosumab or any of the other ingredients of Prolia.
49
2.
Before you use Prolia
Take special care with Prolia
Please tell your doctor immediately if you develop a swollen, red area of skin, most commonly in the
lower leg, that feels hot and tender (cellulitis), and possibly with symptoms of fever while being on
treatment with Prolia.
Tell your doctor if you have or have ever had severe kidney problems, kidney failure or have needed
dialysis.
You should also take calcium and vitamin D supplements while being on treatment with Prolia. Your
doctor will discuss this with you.
A dental examination should be considered before you start treatment with Prolia if you have cancer,
are undergoing chemotherapy or radiotherapy, are taking steroids, do not receive routine dental care or
have gum disease.
If you are under dental treatment or will undergo dental surgery, tell your dentist that you are being
treated with Prolia.
It is important to maintain good oral hygiene when being on treatment with Prolia.
Prolia is not recommended for anyone under 18 years of age. The use of Prolia in children and
adolescents has not been studied.
Using other medicines
Please tell your doctor or pharmacist if you are taking or have recently taken any other medicines,
including medicines obtained without a prescription.
Pregnancy and breast-feeding
Prolia has not been tested in pregnant women. It is important to tell your doctor if you are pregnant;
think you may be pregnant; or plan to get pregnant. Prolia is not recommended for use if you are
pregnant.
It is not known whether Prolia is excreted in breast milk. It is important to tell your doctor if you are
breast-feeding or plan to do so. Your doctor will then help you decide whether to stop breast-feeding,
or whether to stop taking Prolia, considering the benefit of breast-feeding to the baby and the benefit
of Prolia to the mother.
Ask your doctor or pharmacist for advice before taking any medicine.
Driving and using machines
Prolia has no or negligible influence on the ability to drive and use machines
Important information about some of the ingredients of Prolia
If you have an intolerance to some sugars
If you have been told by your doctor that you have an intolerance to some sugars (sorbitol E420),
contact your doctor before taking this medicinal product.
If you are on a controlled sodium diet
This medicinal product contains less than 1 mmol sodium (23 mg) per 60 mg, i.e. essentially ‘sodium-
free’.
50
3.
HOW TO USE PROLIA
The usual dose is 60 mg administered once every 6 months, as a single injection under the skin
(subcutaneous).
The best places to inject are into the thigh, abdomen or
outer area of the upper arm.
Each pack of Prolia contains a reminder card
with stickers that can be removed from the carton. Use the peel-off stickers to
mark the next injection date on your personal calendar and/or the reminder
card to keep a record of the next injection date.
You should also take calcium and vitamin D supplements while being on treatment with Prolia. Your
doctor will discuss this with you.
Your doctor or healthcare provider will show your carer how to use Prolia.
If you forget to use Prolia
If a dose of Prolia is missed, the injection should be administered as soon as possible. Thereafter,
injections should be scheduled every 6 months from the date of the last injection.
If you stop using Prolia
To get the most benefit from your treatment, it is important to use Prolia for as long as your doctor
prescribes it for you. Please talk to your doctor before you consider stopping the treatment.
4.
POSSIBLE SIDE EFFECTS
Uncommonly, patients receiving Prolia may develop skin infections (predominantly cellulitis).
Please
tell your doctor immediately
if you develop any of these symptoms while being on treatment with
Prolia: swollen, red area of skin, most commonly in the lower leg, that feels hot and tender, and
possibly with symptoms of fever.
Like all medicines, Prolia can cause side effects, although not everybody gets them.
The frequency of possible side effects listed below is defined using the following convention:
Very common (affects more than 1 user in 10)
Common (affects 1 to 10 users in 100)
Uncommon (affects 1 to 10 users in 1,000)
Rare (affects 1 to 10 users in 10,000)
Very rare (affects less than 1 user in 10,000)
Not known (frequency cannot be estimated from the available data).
Common side effects
:
•
painful urination, frequent urination, blood in the urine, inability to hold your urine,
•
upper respiratory tract infection,
•
pain, tingling or numbness that moves down your leg (sciatica),
•
cloudy area in the lens of the eye (cataracts),
•
constipation,
•
rash,
51

•
arm or leg pain (pain in extremity).
Uncommon side effects
:
•
swollen, red area of skin, most commonly in the lower leg, that feels hot and tender (cellulitis),
and possibly with symptoms of fever,
•
fever vomiting and abdominal pain or discomfort (diverticulitis),
•
ear infection,
•
skin condition with itching, redness and/or dryness (eczema).
Rare side effects:
•
Persistent pain and/or non-healing sores of the mouth or jaw.
Very rare side effects
:
•
low calcium levels in the blood (hypocalcaemia).
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet, please
tell your doctor or pharmacist.
5.
HOW TO STORE PROLIA
Keep out of the reach and sight of children.
Do not use Prolia after the expiry date which is stated on the label and carton after EXP. The expiry
date refers to the last day of that month.
Store in a refrigerator (2°C – 8°C).
Do not freeze.
Store in the original carton in order to protect from light.
Do not shake excessively.
Your vial may be left outside the refrigerator to reach room temperature (up to 25°C) before injection.
This will make the injection more comfortable. Once your vial has been left to reach room
temperature (up to 25°C), it must be used within 30 days.
Medicines should not be disposed of via wastewater or household waste. Ask your pharmacist how to
dispose of medicines no longer required. These measures will help to protect the environment.
6.
FURTHER INFORMATION
What Prolia contains
-
The active substance is denosumab. Each 1 ml vial contains 60 mg of denosumab (60 mg/ml).
-
The other ingredients are glacial acetic acid, sodium hydroxide, sorbitol (E420) and water for
injections.
What Prolia looks like and contents of the pack
Prolia is a clear, colourless to slightly yellow solution for injection provided in a vial. It may contain
trace
amounts of clear to white particles. Each pack contains one vial.
52
Marketing Authorisation Holder and Manufacturer
Amgen Europe B.V.
Minervum 7061
NL-4817 ZK Breda
The Netherlands
For any information about this medicine, please contact the local representative of the Marketing
Authorisation Holder.
België/Belgique/Belgien
s.a. Amgen n.v.
Tel/Tél: +32 (0)2 7752711
Luxembourg/Luxemburg
s.a. Amgen
Belgique/Belgien
Tel/Tél: +32 (0)2 7752711
България
Амджен България ЕООД
Тел: +359 (0) 2 805 7020
Magyarország
Amgen Kft.
Tel.: +36 1 35 44 700
Česká republika
Amgen s.r.o
Tel : +420 2 21 773 500
Malta
Amgen B.V.
The Netherlands
Tel : +31 (0) 76 5732500
Danmark
Amgen filial af Amgen AB, Sverige
Tlf: +45 39617500
Nederland
Amgen B.V.
Tel: +31 (0) 76 5732500
Deutschland
AMGEN GmbH
Tel: +49 (0)89 1490960
Norge
Amgen AB
Tel: +47 23308000
Eesti
Amgen Switzerland AG Eesti filiaal
Tel: +372 5125 501
Österreich
Amgen GmbH
Tel: +43 (0) 1 50 217
Ελλάδα
Amgen Ελλάς Φαρμακευτικά ΕΠΕ.
Τηλ.: +30 210 3447000
Polska
Amgen Sp. z o.o.
Tel.: +48 22 581 3000
España
Amgen S.A.
Tel: +34 93 600 19 00
Portugal
AMGEN Biofarmacêutica, Lda.
Tel: +351 21 4220550
France
Amgen S.A.S
Tél: +33 (0)1 40 88 27 00
România
Mediplus Exim SRL
Tel.: +4021 301 74 74
Ireland
Amgen Limited
United Kingdom
Tel:
+44 (0)1223 420305
Slovenija
AMGEN zdravila d.o.o.
Tel: +386 1 585 1767
53
Ísland
Vistor hf.
Sími: +354 535 7000
Slovenská republika
Amgen Switzerland AG Slovakia
Tel : +421 33 321 13 22
Italia
Amgen Dompé S.p.A.
Tel: +39 02 6241121
Suomi/Finland
Amgen AB, sivuliike Suomessa/Amgen AB, filial
i Finland
Puh/Tel: +358 (0)9 54900500
Kύπρος
Papaellinas & Co Ltd
Τηλ.: + 357 22741 741
Sverige
Amgen AB
Tel: +46 (0)8 6951100
Latvija
Amgen Switzerland AG Rīgas filiāle
Tel : +371 29284 807
United Kingdom
Amgen Limited
Tel: +44 (0)1223 420305
Lietuva
Amgen Switzerland AG Vilniaus filialas
Tel. +370 6983 6600
This leaflet was last approved in
{MM/YYYY}.
Detailed information on this medicine is available on the European Medicines Agency web site:
http://www.ema.europa.eu/
54
Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/prolia.html
Copyright © 1995-2021 ITA all rights reserved.