










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
ReFacto AF 250 IU powder and solvent for solution for injection.
2.
Each vial contains nominally 250 IU* moroctocog alfa**.
After reconstitution, each ml of solution contains approximately 62.5 IU moroctocog alfa.
* The potency (International Units) is determined using the European Pharmacopoeia chromogenic
assay. The specific activity of ReFacto AF is 7,600-13,800 IU/mg protein.
** Human coagulation factor VIII produced by recombinant DNA technology in Chinese hamster
ovary (CHO) cells. Moroctocog alfa is a glycoprotein with 1438 amino acids with a sequence that is
comparable to the 90 + 80 kDa form of factor VIII (i.e. B-domain deleted) and similar
post-translational modifications to those of the plasma-derived molecule.
The manufacturing process for ReFacto has been modified to eliminate any exogenous human- or
animal-derived protein in the cell culture process, purification, or final formulation; and at the same
time the invented name has been changed to ReFacto AF.
Excipients:
After reconstitution, 1.23 mmol (29 mg) sodium per vial.
For a full list of excipients, see section 6.1.
3.
Powder and solvent for solution for injection.
White to off-white cake/powder.
Clear, colourless solvent.
4.
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor VIII
deficiency).
ReFacto AF is appropriate for use in adults and children of all ages, including newborns.
ReFacto AF does not contain von Willebrand factor, and hence is not indicated in von Willebrand’s
disease.
Treatment should be initiated under the supervision of a physician experienced in the treatment of
haemophilia A.
Posology
2
The labelled potency of ReFacto AF is based on the European Pharmacopoeial chromogenic substrate
assay, in which the manufacturing potency standard has been calibrated to the WHO International
Standard using the chromogenic substrate assay. When monitoring patients' factor VIII activity levels
during treatment with ReFacto AF, use of the European Pharmacopoeial chromogenic substrate assay
is strongly recommended. The chromogenic assay yields results which are higher than those observed
with use of the one-stage clotting assay.
Typically, one-stage clotting assay results are 20-50% lower
than the chromogenic substrate assay results.
The ReFacto AF laboratory standard can be used to
correct for this discrepancy (see
section 5.2)
.
Another moroctocog alfa product approved for use outside Europe has a different potency assigned
using a manufacturing potency standard that has been calibrated to the WHO International Standard
using a one-stage clotting assay; this product is identified by the tradename XYNTHA. Due to the
difference in methods used to assign product potency of XYNTHA and ReFacto AF, 1 IU of the
XYNTHA product (one-stage assay calibrated) is approximately equivalent to 1.38 IU of the ReFacto
AF product (chromogenic assay calibrated). If a patient normally treated with XYNTHA is prescribed
ReFacto AF, the treating physician may consider adjustment of dosing recommendations based on
factor VIII recovery values.
Based on their current regimen, individuals with haemophilia A should be advised to bring an
adequate supply of factor VIII product for anticipated treatment when travelling. Patients should be
advised to consult with their healthcare provider prior to travel.
The dosage and duration of the substitution therapy depend on the severity of the factor VIII
deficiency, on the location and extent of bleeding, and on the patient’s clinical condition. Doses
administered should be titrated to the patient's clinical response. In the presence of an inhibitor, higher
doses or appropriate specific treatment may be required.
The number of units of factor VIII administered is expressed in International Units (IUs), which are
related to the current WHO standard for factor VIII products. Factor VIII activity in plasma is
expressed either as a percentage (relative to normal human plasma) or in International Units (relative
to an International Standard for factor VIII in plasma). One International Unit (IU) of factor VIII
activity is equivalent to the quantity of factor VIII in one ml of normal human plasma. The calculation
of the required dosage of factor VIII is based upon the empirical finding that 1 International Unit (IU)
of factor VIII per kg body weight raises the plasma factor VIII activity by 2 IU/dl. The required
dosage is determined using the following formula:
Required units (IU) = body weight (kg) x desired factor VIII rise (% or IU/dl) x 0.5 (IU/kg per IU/dl),
where 0.5 IU/kg per IU/dl represents the reciprocal of the incremental recovery generally observed
following infusions of factor VIII.
The amount to be administered and the frequency of administration should always be oriented to the
clinical effectiveness in the individual case.
In the case of the following haemorrhagic events, the factor VIII activity should not fall below the
given plasma levels (in % of normal or in IU/dl) in the corresponding period. The following table can
be used to guide dosing in bleeding episodes and surgery:
3
Degree of haemorrhage/
Type of surgical procedure
Factor VIII level
required (% or IU/dl)
Frequency of doses (hours)/
Duration of therapy (days)
Haemorrhage
Early haemarthrosis, muscle
bleeding or oral bleeding
20-40
Repeat every 12-24 hours. At least 1
day until the bleeding episode as
indicated by pain is resolved or healing
is achieved.
More extensive haemarthrosis,
muscle bleeding or haematoma
30-60
Repeat infusion every 12-24 hours for
3-4 days or more until pain and acute
disability are resolved.
Life-threatening haemorrhages
60-100
Repeat infusion every 8-24 hours until
threat is resolved.
Surgery
Minor,
including tooth extraction
30-60
Every 24 hours, at least 1 day, until
healing is achieved.
Major
80-100
(pre- and
post-operative)
Repeat infusion every 8-24 hours until
adequate wound healing, then therapy
for at least another 7 days to maintain a
factor VIII activity of 30% to 60%
(IU/dl).
During the course of treatment, appropriate determination of factor VIII levels is advised to guide the
dose to be administered and the frequency of repeated infusions. In the case of major surgical
interventions in particular, precise monitoring of the substitution therapy by means of coagulation
analysis (plasma factor VIII activity) is indispensable. Individual patients may vary in their response
to factor VIII, achieving different levels of
in vivo
recovery and demonstrating different half-lives.
For long-term prophylaxis against bleeding in patients with severe haemophilia A, the usual doses are
20 to 40 IU of factor VIII per kg body weight at intervals of 2 to 3 days. In some cases, especially in
younger patients, shorter dosage intervals or higher doses may be necessary.
Patients using factor-VIII replacement therapy are to be monitored for the development of factor VIII
inhibitors. If expected factor VIII activity plasma levels are not attained, or if bleeding is not
controlled with an appropriate dose, an assay should be performed to determine if factor VIII
inhibitors are present. Data from clinical trials indicated that if inhibitors are present at levels less than
10 Bethesda Units (BUs), administration of additional antihaemophilic factor may neutralise the
inhibitors. In patients with levels of inhibitor above 10 BU, factor VIII therapy may not be effective
and other therapeutic options should be considered. Management of such patients should be directed
by physicians with experience in the care of patients with haemophilia (see
section 4.4)
.
Special populations
Renal or hepatic impairment
Dosage adjustment for patients with renal or hepatic impairment has not been studied in clinical trials.
4


Paediatric patients
Safety and efficacy studies with ReFacto have been performed both in previously treated children and
adolescents (n=31, ages 8-18 years) and in previously untreated neonates, infants and children (n=101,
ages < 1-52 months).
The need for an increased dose relative to that used for adults and older children should be anticipated
when treating younger children with ReFacto AF. In a study of ReFacto in children less than 6 years
of age, pharmacokinetic analysis revealed half-life and recovery less than that observed in older
children and adults (see section 5.2). During the clinical trials, children less than 6 years of age on a
prophylaxis regimen used an average dose of 50 IU/kg of ReFacto and experienced an average of
6.1 bleeding episodes per year. Older children and adults on a prophylaxis regimen used an average
dose of 27 IU/kg and experienced an average of 10 bleeding episodes per year. In a clinical trial
setting the mean dose per infusion of ReFacto for bleeding episodes in children less than 6 years of
age was higher than the mean dose administered to older children and adults (51.3 IU/kg and
29.3 IU/kg, respectively).
Method of administration
ReFacto AF is administered by intravenous injection over several minutes after reconstitution of the
lyophilised powder for injection with sodium chloride 9 mg/ml (0.9%) solution for injection
(provided). The rate of administration should be determined by the patient’s comfort level.
Appropriate training is recommended for non-healthcare professionals administering the product.
In the interest of patients, it is recommended that every time ReFacto AF is administered, the name
and batch number of the product should be recorded.
For reconstitution instructions prior to administration, see section 6.6.
Hypersensitivity to the active substance or to any of the excipients.
Hypersensitivity to hamster proteins.
As with any intravenous protein product, allergic-type hypersensitivity reactions are possible. The
product contains traces of hamster proteins. Patients should be informed of the early signs of
hypersensitivity reactions (including hives, generalised urticaria, tightness of the chest, wheezing,
hypotension) and anaphylaxis. If allergic or anaphylactic reactions occur, administration of ReFacto
AF is to be discontinued immediately, and an appropriate treatment must be initiated. In case of shock,
the current medical standards for treatment of shock are to be observed. Patients are to be advised to
discontinue use of the product and contact their physician or seek immediate emergency care,
depending on the type and severity of the reaction, if any of these symptoms occur.
The formation of neutralising antibodies (inhibitors) to factor VIII is a known complication in the
management of individuals with haemophilia A. These inhibitors are usually IgG immunoglobulins
directed against the factor VIII procoagulant activity, which are quantified in Bethesda Units (BUs)
per ml of plasma using the Nijmegen modification of the Bethesda assay. The risk of developing
inhibitors is correlated to the exposure to factor VIII, this risk being highest within the first
20 exposure days. Inhibitors have been observed in previously treated patients receiving factor VIII
products, including ReFacto AF. Cases of recurrence of inhibitors (low titre) have been observed after
switching from one recombinant factor VIII product to another in previously treated patients with
more than 100 exposure days who have a history of inhibitor development. Patients treated with
5
recombinant coagulation factor VIII should be carefully monitored for the development of inhibitors
by appropriate clinical observations and laboratory tests (see also
section 4.8)
.
Reports of lack of effect, mainly in prophylaxis patients, have been received in the clinical trials and in
the post-marketing setting for ReFacto. The reported lack of effect with ReFacto has been described as
bleeding into target joints, bleeding into new joints or a subjective feeling by the patient of new onset
bleeding. When prescribing ReFacto AF it is important to individually titrate and monitor each
patient's factor level in order to ensure an adequate therapeutic response.
In the interest of patient safety, it is recommended that every time ReFacto AF is administered, the
name on the carton and batch number of the product are recorded. Patients can affix one of the peel-
off labels found on the vial to document the batch number in their diary or for reporting any side
effects.
After reconstitution this medicinal product contains 1.23 mmol (29 mg) sodium per vial, to be taken
into consideration by patients on a controlled sodium diet.
No interaction studies have been performed.
Animal reproduction studies have not been conducted with factor VIII. Because of the rare occurrence
of haemophilia A in women, experience regarding the use of factor VIII during pregnancy and breast-
feeding is not available. Therefore, factor VIII should be used during pregnancy and lactation only if
clearly indicated.
No studies on the effects on the ability to drive and use machines have been performed.
Factor VIII inhibition
The occurrence of neutralising antibodies (inhibitors) to factor VIII is well known in the treatment of
patients with haemophilia A. As with all coagulation factor VIII products, patients are to be monitored
for the development of inhibitors that are to be titrated in Bethesda Units (BUs) using the Nijmegen
modification of the Bethesda assay. If such inhibitors occur, the condition may manifest itself as an
insufficient clinical response. In such cases, it is recommended that a specialised haemophilia centre
be contacted.
In a clinical study with ReFacto AF in previously treated patients (PTPs), the incidence of factor VIII
inhibitors was the primary safety endpoint. Two clinically silent, low-titre, transient inhibitors were
observed in 94 patients with a median exposure of 76 exposure days (ED, range 1-92), corresponding
to 2.2% of the 89 patients with at least 50 ED. In a supporting study of ReFacto AF, 1
de novo
and 2
recurrent inhibitors (all low-titre, central laboratory determination) were observed in 110 patients;
median exposure of 58 ED (range 5-140) and 98 patients had at least 50 ED to ReFacto AF.
Ninety-eight (98) of the original 110 patients continued treatment in a second supportive study and
had subsequent extended exposure to ReFacto AF with a median of 169 additional ED (range 9-425).
One (1) additional low-titre
de novo
inhibitor was observed. The frequency of inhibitors observed in
these studies is within the expected range.
In a clinical study with ReFacto in PTPs, 1 inhibitor was observed in 113 patients. Also, there have
been spontaneous post-marketing reports of high-titre inhibitors involving previously treated patients.
6
There are no clinical data on previously untreated patients (PUPs) with ReFacto AF. However, clinical
trials are planned in previously untreated patients (PUPs) with ReFacto AF. In a clinical trial, 32 out of
101 (32%) previously untreated patients (PUPs) treated with ReFacto developed inhibitors: 16 out of
101 (16%) with a titre > 5 BU and 16 out of 101 (16%) with a titre ≤ 5 BU. The median number of
exposure days up to inhibitor development in these patients was 12 (range 3-49). Of the 16 patients
with high titres, 15 received immune tolerance (IT) treatment. Of the 16 patients with low titres, IT
treatment was started in 10. IT had an efficacy of 73% for patients with high titres and 90% for those
with low titres. For all 101 treated PUPs, regardless of inhibitor development, the median number of
exposure days is 197 (range 1-1299).
Adverse reactions based on experience from clinical trials with ReFacto or ReFacto AF are presented
in the table below by system organ class. These frequencies have been estimated on a per-patient basis
and are described using the following categories: very common (1/10); common (1/100 to <1/10);
and uncommon (1/1,000 to <1/100).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Frequency of Occurrence per Patient with ReFacto or ReFacto AF
System Organ Class
Very common
(≥1/10)
Common
(1/100 to <1/10)
Uncommon
(1/1,000 to <1/100)
Blood and lymphatic
disorders
Factor VIII
inhibitors - PUPs
Factor VIII inhibitors -
PTPs
Metabolism and
nutrition disorders
Anorexia
Nervous system
disorders
Headache
Neuropathy, dizziness,
somnolence, dysgeusia
Cardiac disorders
Angina pectoris, tachycardia,
palpitations
Vascular disorders
Haemorrhage/
Haematoma
Hypotension, thrombophlebitis,
vasodilatation, flushing
Respiratory, thoracic
and mediastinal
disorders
Dyspnoea, cough
Gastrointestinal
disorders
Vomiting
Nausea
Abdominal pain, diarrhoea
Skin and subcutaneous
tissue disorders
Urticaria, pruritis, rash,
hyperhidrosis
Musculoskeletal,
connective tissue and
bone disorders
Arthralgia
Myalgia
General disorders and
administration site
conditions
Asthenia, pyrexia
Chills/feeling cold, injection
site inflammation, injection site
reaction, injection site pain
Investigations
Aspartate aminotransferase
increased, alanine
aminotransferase increased,
blood bilirubin increased, blood
creatine phosphokinase
increased
Surgical and medical
procedures
Vascular access
complication
One event of cyst in an 11-year old patient and one event described as confusion in a 13-year old
patient have been reported as possibly related to ReFacto AF treatment.
7




















Safety of ReFacto AF was evaluated in previously treated children and adolescents (n=18, age 12-16
in a study and n=49, age 7-16 in a supporting study). Although a limited number of children have been
studied, there is a tendency for higher frequencies of adverse events in children aged 7-16 as compared
to adults. A clinical trial evaluating use of moroctocog alfa (AF-CC) in children less than 6 years of
age is on going.
The following adverse events have also been reported for ReFacto: paraesthesia, fatigue, blurred
vision, acne, gastritis, gastroenteritis, and pain.
Hypersensitivity or allergic reactions (which may include angioedema, burning and stinging at the
infusion site, chills, flushing, generalised urticaria, headache, hives, hypotension, lethargy, nausea,
restlessness, tachycardia, tightness of the chest, tingling, vomiting, wheezing) have been observed
infrequently for ReFacto, and may in some cases progress to severe anaphylaxis including shock (see
Trace amounts of hamster protein may be present in ReFacto AF. Very rarely, development of
antibodies to hamster protein has been observed, but there were no clinical sequelae. In a study of
ReFacto, twenty of 113 (18%) PTPs had an increase in anti-CHO antibody titre, without any apparent
clinical effect.
If any reaction takes place that is thought to be related to the administration of ReFacto AF, the rate of
infusion is to be decreased or the infusion stopped, as dictated by the response of the patient (see
No case of overdose has been reported.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antihaemorrhagics, blood coagulation factor VIII; ATC code: B02BD02.
ReFacto AF contains B-domain deleted recombinant coagulation factor VIII (moroctocog alfa). It is a
glycoprotein with an approximate molecular mass of 170,000 Da consisting of 1438 amino acids.
ReFacto AF has functional characteristics comparable to those of endogenous factor VIII. Factor VIII
activity is greatly reduced in patients with haemophilia A, and, therefore, replacement therapy is
necessary.
When infused into a haemophiliac patient, factor VIII binds to the von Willebrand factor present in the
patient’s circulation.
Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X
to activated factor X. Activated factor X converts prothrombin into thrombin. Thrombin then converts
fibrinogen into fibrin, and a clot is formed. Haemophilia A is a sex-linked hereditary disorder of blood
coagulation due to decreased levels of factor VIII:C and results in profuse bleeding into joints,
muscles or internal organs, either spontaneously or as a result of accidental or surgical trauma. By
replacement therapy, the plasma levels of factor VIII are increased, thereby enabling a temporary
correction of the factor deficiency and correction of the bleeding tendencies.
8
5.2 Pharmacokinetic properties
Pharmacokinetic properties of ReFacto, derived from a cross-over study of ReFacto and a plasma-
derived FVIII concentrate, using the chromogenic substrate assay (see section 4.2), in 18 previously
treated patients are listed in the table below.
Pharmacokinetic parameter estimates for ReFacto in previously treated patients with haemophilia A
PK parameter
Mean
SD
Median
AUC
t
(IU·h/ml)
19.9
4.9
19.9
t
1/2
(h)
14.8
5.6
12.7
CL (ml/h·kg)
2.4
0.75
2.3
MRT (h)
20.2
7.4
18.0
K-value
(IU/dl increase in FVIII:C per IU/kg FVIII given) 2.4 0.38 2.5
Abbreviations: AUC
t
= area under the plasma concentration-time curve from zero to the last measurable
concentration; t
½
= half-life; CL = clearance; MRT = mean residence time; K-value = incremental recovery;
SD = standard deviation
In a study in which the potency of ReFacto AF, ReFacto and FVIII activity in patient plasma were
measured using the chromogenic substrate assay, ReFacto AF was shown to be bioequivalent to
ReFacto. The ratios of geometric least-square means of ReFacto AF-to-ReFacto were 100.6%, 99.5%
and 98.1% for K-value, AUC
t
and AUC
∞
(area under the plasma concentration curve from time zero
to infinity), respectively. The corresponding 90% confidence intervals about the ratios of ReFacto AF
to ReFacto geometric means were within the bioequivalence window of 80% to 125%, demonstratin
bioequivalence of ReFacto AF to ReFacto.
g
In a cross-over pharmacokinetic study, the pharmacokinetic parameters for ReFacto AF were
determined at baseline and followed-up in 25 previously treated patients (≥ 12 years) after repeated
administration of ReFacto AF for six months. The ratios of geometric least-square means of month
6-to-baseline pharmacokinetic were 107%, 100% and 104% for K-value, AUC
t
and AUC
,
respectively. The corresponding 90% confidence intervals about the ratios of month 6-to-baseline for
the above pharmacokinetic parameters were within the equivalence window of 80% to 125%. This
indicates no time-dependent changes in the pharmacokinetic properties of ReFacto AF.
In the same study, in which the drug potency of ReFacto AF and a full-length recombinant factor VIII
(FLrFVIII) comparator, and the FVIII activity measured in patient plasma samples were all
determined using the same one-stage clotting assay at a central laboratory, ReFacto AF was shown to
be pharmacokinetically equivalent to FLrFVIII in 30 previously treated patients (≥ 12 years) using the
standard bioequivalence approach.
In PUPs, pharmacokinetic parameters of ReFacto were evaluated using the chromogenic assay. These
patients (n=59; median age 10 ± 8.3 months) had a mean incremental recovery at Week 0 of
1.5 ± 0.6 IU/dl per IU/kg (range 0.2 to 2.8 IU/dl per IU/kg) which was lower than that obtained in
PTPs treated with ReFacto at Week 0 with a mean K-value of 2.4 ± 0.4 IU/dl per IU/kg (range 1.1 to
3.8 IU/dl per IU/kg). In the PUPs, the mean incremental recovery was stable over time (5 visits during
a 2-year period) and ranged from 1.5 to 1.8 IU/dl per IU/kg. Population pharmacokinetic modeling
using data from 44 PUPs led to a mean estimated half-life of 8.0 ± 2.2 hours.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity, and genotoxicity.
No investigations on carcinogenic potential or toxicity to reproduction have been conducted.
9










6.
6.1 List of excipients
Powder
Sucrose
Calcium chloride dihydrate
L-Histidine
Polysorbate 80
Sodium chloride
Solvent
Sodium chloride
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products, including other infusion solutions.
Only the provided infusion set is to be used, because treatment failure can occur as a consequence of
human-coagulation factor VIII adsorption to the internal surfaces of some infusion equipment.
6.3 Shelf life
Unopened powder vial
3 years.
After reconstitution
Chemical and physical in-use stability has been demonstrated for 3 hours at temperatures up to 25
o
C.
The product does not contain a preservative, and the reconstituted product should be used
immediately, or within 3 hours after reconstitution. Other in-use storage times and conditions are the
responsibility of the user.
6.4 Special precautions for storage
Store and transport refrigerated (2°C - 8°C). Do not freeze, in order to prevent damage to the pre-filled
syringe.
The product may be removed from refrigerated storage for one single period of maximum 3 months at
room temperature (up to 25°C). At the end of this period of room temperature storage, the product
must not be returned to refrigerated storage, but is to be used or discarded.
Keep the vial in the outer carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
10
6.5 Nature and contents of container
250 IU powder in a 10 ml vial (type 1 glass) with a stopper (butyl) and a flip-off seal (aluminum) and
4 ml of solvent in a pre-filled syringe (type 1 glass) with a plunger stopper (butyl), a tip-cap (butyl)
and a sterile vial adapter reconstitution device, a sterile infusion set, alcohol swabs, a plaster and a
gauze pad.
6.6
Special precautions for disposal and other handling
The vial of lyophilised product powder for injection must be reconstituted with the supplied solvent
[sodium chloride 9 mg/ml (0.9%) solution] from the pre-filled syringe using the sterile vial adapter
reconstitution device. The vial should be gently rotated until all of the powder is dissolved.
The product, when reconstituted, contains polysorbate-80, which is known to increase the rate of di-
(2-ethylhexyl)phthalate (DEHP) extraction from polyvinyl chloride (PVC). This is to be considered
during the preparation and administration of the product, including storage time elapsed in a PVC
container following reconstitution. It is important that the recommendations in section 6.3 be followed
closely.
After reconstitution, the solution is drawn back into the syringe. The solution will be clear or slightly
opalescent and colourless. The solution is to be discarded if visible particulate matter or discolouration
is observed.
Any unused product or waste material is to be disposed of in accordance with local requirements.
7.
Wyeth Europa Ltd.
Huntercombe Lane South
Taplow, Maidenhead
Berkshire, SL6 0PH
United Kingdom
8.
EU/1/99/103/001
9.
Date of first authorisation: 13 April 1999
Date of last renewal: 15 April 2009
Detailed information on this medicinal product is available on the website of the European Medicines
Agency (EMEA) http://www.emea.europa.eu/.
11
1.
ReFacto AF 500 IU powder and solvent for solution for injection.
2.
Each vial contains nominally 500 IU* moroctocog alfa**.
After reconstitution, each ml of solution contains approximately 125 IU moroctocog alfa.
* The potency (International Units) is determined using the European Pharmacopoeia chromogenic
assay. The specific activity of ReFacto AF is 7,600-13,800 IU/mg protein.
** Human coagulation factor VIII produced by recombinant DNA technology in Chinese hamster
ovary (CHO) cells. Moroctocog alfa is a glycoprotein with 1438 amino acids with a sequence that is
comparable to the 90 + 80 kDa form of factor VIII (i.e. B-domain deleted) and similar post-
translational modifications to those of the plasma-derived molecule.
The manufacturing process for ReFacto has been modified to eliminate exogenous human- or animal-
derived protein in the cell culture process, purification, or final formulation; and at the same time the
invented name has been changed to ReFacto AF.
Excipients:
After reconstitution, 1.23 mmol (29 mg) sodium per vial.
For a full list of excipients, see section 6.1.
3.
Powder and solvent for solution for injection.
White to off-white cake/powder.
Clear, colourless solvent.
4.
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor VIII
deficiency).
ReFacto AF is appropriate for use in adults and children of all ages, including newborns.
ReFacto AF does not contain von Willebrand factor, and hence is not indicated in von Willebrand’s
disease.
Treatment should be initiated under the supervision of a physician experienced in the treatment of
haemophilia A.
Posology
12
The labelled potency of ReFacto AF is based on the European Pharmacopoeial chromogenic substrate
assay, in which the manufacturing potency standard has been calibrated to the WHO International
Standard using the chromogenic substrate assay. When monitoring patients' factor VIII activity levels
during treatment with ReFacto AF, use of the European Pharmacopoeial chromogenic substrate assay
is strongly recommended. The chromogenic assay yields results which are higher than those observed
with use of the one-stage clotting assay.
Typically, one-stage clotting assay results are 20-50% lower
than the chromogenic substrate assay results.
The ReFacto AF laboratory standard can be used to
correct for this discrepancy (see
section 5.2)
.
Another moroctocog alfa product approved for use outside Europe has a different potency assigned
using a manufacturing potency standard that has been calibrated to the WHO International Standard
using a one-stage clotting assay; this product is identified by the tradename XYNTHA. Due to the
difference in methods used to assign product potency of XYNTHA and ReFacto AF, 1 IU of the
XYNTHA product (one-stage assay calibrated) is approximately equivalent to 1.38 IU of the ReFacto
AF product (chromogenic assay calibrated). If a patient normally treated with XYNTHA is prescribed
ReFacto AF, the treating physician may consider adjustment of dosing recommendations based on
factor VIII recovery values.
Based on their current regimen, individuals with haemophilia A should be advised to bring an
adequate supply of factor VIII product for anticipated treatment when travelling. Patients should be
advised to consult with their healthcare provider prior to travel.
The dosage and duration of the substitution therapy depend on the severity of the factor VIII
deficiency, on the location and extent of bleeding, and on the patient’s clinical condition. Doses
administered should be titrated to the patient's clinical response. In the presence of an inhibitor, higher
doses or appropriate specific treatment may be required.
The number of units of factor VIII administered is expressed in International Units (IUs), which are
related to the current WHO standard for factor VIII products. Factor VIII activity in plasma is
expressed either as a percentage (relative to normal human plasma) or in International Units (relative
to an International Standard for factor VIII in plasma). One International Unit (IU) of factor VIII
activity is equivalent to the quantity of factor VIII in one ml of normal human plasma. The calculation
of the required dosage of factor VIII is based upon the empirical finding that 1 International Unit (IU)
of factor VIII per kg body weight raises the plasma factor VIII activity by 2 IU/dl. The required
dosage is determined using the following formula:
Required units (IU) = body weight (kg) x desired factor VIII rise (% or IU/dl) x 0.5 (IU/kg per IU/dl),
where 0.5 IU/kg per IU/dl represents the reciprocal of the incremental recovery generally observed
following infusions of factor VIII.
The amount to be administered and the frequency of administration should always be oriented to the
clinical effectiveness in the individual case.
In the case of the following haemorrhagic events, the factor VIII activity should not fall below the
given plasma levels (in % of normal or in IU/dl) in the corresponding period. The following table can
be used to guide dosing in bleeding episodes and surgery:
13
Degree of haemorrhage/
Type of surgical procedure
Factor VIII level
required (% or IU/dl)
Frequency of doses (hours)/
Duration of therapy (days)
Haemorrhage
Early haemarthrosis, muscle
bleeding or oral bleeding
20-40
Repeat every 12-24 hours. At least 1
day until the bleeding episode as
indicated by pain is resolved or healing
is achieved.
More extensive haemarthrosis,
muscle bleeding or haematoma
30-60
Repeat infusion every 12-24 hours for
3-4 days or more until pain and acute
disability are resolved.
Life-threatening haemorrhages
60-100
Repeat infusion every 8-24 hours until
threat is resolved.
Surgery
Minor,
including tooth extraction
30-60
Every 24 hours, at least 1 day, until
healing is achieved.
Major
80-100
(pre- and
post-operative)
Repeat infusion every 8-24 hours until
adequate wound healing, then therapy
for at least another 7 days to maintain a
factor VIII activity of 30% to 60%
(IU/dl).
During the course of treatment, appropriate determination of factor VIII levels is advised to guide the
dose to be administered and the frequency of repeated infusions. In the case of major surgical
interventions in particular, precise monitoring of the substitution therapy by means of coagulation
analysis (plasma factor VIII activity) is indispensable. Individual patients may vary in their response
to factor VIII, achieving different levels of
in vivo
recovery and demonstrating different half-lives.
For long-term prophylaxis against bleeding in patients with severe haemophilia A, the usual doses are
20 to 40 IU of factor VIII per kg body weight at intervals of 2 to 3 days. In some cases, especially in
younger patients, shorter dosage intervals or higher doses may be necessary.
Patients using factor VIII replacement therapy are to be monitored for the development of factor VIII
inhibitors. If expected factor VIII activity plasma levels are not attained, or if bleeding is not
controlled with an appropriate dose, an assay should be performed to determine if factor VIII
inhibitors are present. Data from clinical trials indicated that if inhibitors are present at levels less than
10 Bethesda Units (BUs), administration of additional antihaemophilic factor may neutralise the
inhibitors. In patients with levels of inhibitor above 10 BU, factor VIII therapy may not be effective
and other therapeutic options should be considered. Management of such patients should be directed
by physicians with experience in the care of patients with haemophilia (see
section 4.4)
.
Special populations
Renal or hepatic impairment
Dosage adjustment for patients with renal or hepatic impairment has not been studied in clinical trials.
Paediatric patients
Safety and efficacy studies with ReFacto have been performed both in previously treated children and
adolescents (n=31, ages 8-18 years) and in previously untreated neonates, infants and children (n=101,
ages < 1-52 months).
14


The need for an increased dose relative to that used for adults and older children should be anticipated
when treating younger children with ReFacto AF. In a study of ReFacto in children less than 6 years
of age, pharmacokinetic analysis revealed half-life and recovery less than that observed in older
children and adults (see section 5.2). During the clinical trials, children less than 6 years of age on a
prophylaxis regimen used an average dose of 50 IU/kg of ReFacto and experienced an average of
6.1 bleeding episodes per year. Older children and adults on a prophylaxis regimen used an average
dose of 27 IU/kg and experienced an average of 10 bleeding episodes per year. In a clinical trial
setting the mean dose per infusion of ReFacto for bleeding episodes in children less than 6 years of
age was higher than the mean dose administered to older children and adults (51.3 IU/kg and
29.3 IU/kg, respectively).
Method of administration
ReFacto AF is administered by intravenous injection over several minutes after reconstitution of the
lyophilised powder for injection with sodium chloride 9 mg/ml (0.9%) solution for injection
(provided). The rate of administration should be determined by the patient’s comfort level.
Appropriate training is recommended for non-healthcare professionals administering the product.
In the interest of patients, it is recommended that every time ReFacto AF is administered, the name
and batch number of the product should be recorded.
For reconstitution instructions prior to administration, see section 6.6.
Hypersensitivity to the active substance or to any of the excipients.
Hypersensitivity to hamster proteins.
As with any intravenous protein product, allergic-type hypersensitivity reactions are possible. The
product contains traces of hamster proteins. Patients should be informed of the early signs of
hypersensitivity reactions (including hives, generalised urticaria, tightness of the chest, wheezing,
hypotension) and anaphylaxis. If allergic or anaphylactic reactions occur, administration of ReFacto
AF is to be discontinued immediately, and an appropriate treatment must be initiated. In case of shock,
the current medical standards for treatment of shock are to be observed. Patients are to be advised to
discontinue use of the product and contact their physician or seek immediate emergency care,
depending on the type and severity of the reaction, if any of these symptoms occur.
The formation of neutralising antibodies (inhibitors) to factor VIII is a known complication in the
management of individuals with haemophilia A. These inhibitors are usually IgG immunoglobulins
directed against the factor VIII procoagulant activity, which are quantified in Bethesda Units (BUs)
per ml of plasma using the Nijmegen modification of the Bethesda assay. The risk of developing
inhibitors is correlated to the exposure to factor VIII, this risk being highest within the first
20 exposure days. Inhibitors have been observed in previously treated patients receiving factor VIII
products, including ReFacto AF. Cases of recurrence of inhibitors (low titre) have been observed after
switching from one recombinant factor VIII product to another in previously treated patients with
more than 100 exposure days who have a history of inhibitor development. Patients treated with
recombinant coagulation factor VIII should be carefully monitored for the development of inhibitors
by appropriate clinical observations and laboratory tests (see also
section 4.8)
.
Reports of lack of effect, mainly in prophylaxis patients, have been received in the clinical trials and in
the post-marketing setting for ReFacto. The reported lack of effect with ReFacto has been described as
bleeding into target joints, bleeding into new joints or a subjective feeling by the patient of new onset
15
bleeding. When prescribing ReFacto AF it is important to individually titrate and monitor each
patient’s factor level in order to ensure an adequate therapeutic response.
In the interest of patient safety, it is recommended that every time ReFacto AF is administered, the
name on the carton and batch number of the product are recorded. Patients can affix one of the peel-
off labels found on the vial to document the batch number in their diary or for reporting any side
effects.
After reconstitution this medicinal product contains 1.23 mmol (29 mg) sodium per vial, to be taken
into consideration by patients on a controlled sodium diet.
No interaction studies have been performed.
Animal reproduction studies have not been conducted with factor VIII. Because of the rare occurrence
of haemophilia A in women, experience regarding the use of factor VIII during pregnancy and breast-
feeding is not available. Therefore, factor VIII should be used during pregnancy and lactation only if
clearly indicated.
No studies on the effects on the ability to drive and use machines have been performed.
Factor VIII inhibition
The occurrence of neutralising antibodies (inhibitors) to factor VIII is well known in the treatment of
patients with haemophilia A. As with all coagulation factor VIII products, patients are to be monitored
for the development of inhibitors that are to be titrated in Bethesda Units (BUs) using the Nijmegen
modification of the Bethesda assay. If such inhibitors occur, the condition may manifest itself as an
insufficient clinical response. In such cases, it is recommended that a specialised haemophilia centre
be contacted.
In a clinical study with ReFacto AF in previously treated patients (PTPs), the incidence of factor VIII
inhibitors was the primary safety endpoint. Two clinically silent, low-titre, transient inhibitors were
observed in 94 patients with a median exposure of 76 exposure days (ED, range 1-92), corresponding
to 2.2% of the 89 patients with at least 50 ED. In a supporting study of ReFacto AF, 1
de novo
and 2
recurrent inhibitors (all low-titre, central laboratory determination) were observed in 110 patients;
median exposure of 58 ED (range 5-140) and 98 patients had at least 50 ED to ReFacto AF.
Ninety-eight (98) of the original 110 patients continued treatment in a second supportive study and
had subsequent extended exposure to ReFacto AF with a median of 169 additional ED (range 9-425).
One (1) additional low-titre
de novo
inhibitor was observed. The frequency of inhibitors observed in
these studies is within the expected range.
In a clinical study with ReFacto in PTPs, 1 inhibitor was observed in 113 patients. Also, there have
been spontaneous post-marketing reports of high-titre inhibitors involving previously treated patients.
There are no clinical data on previously untreated patients (PUPs) with ReFacto AF. However, clinical
trials are planned in previously untreated patients (PUPs) with ReFacto AF. In a clinical trial, 32 out of
101 (32%) previously untreated patients (PUPs) treated with ReFacto developed inhibitors: 16 out of
101 (16%) with a titre > 5 BU and 16 out of 101 (16%) with a titre ≤ 5 BU. The median number of
exposure days up to inhibitor development in these patients was 12 (range 3-49). Of the 16 patients
with high titres, 15 received immune tolerance (IT) treatment. Of the 16 patients with low titres, IT
treatment was started in 10. IT had an efficacy of 73% for patients with high titres and 90% for those
16
with low titres. For all 101 treated PUPs, regardless of inhibitor development, the median number of
exposure days is 197 (range 1-1299).
Adverse reactions based on experience from clinical trials with ReFacto or ReFacto AF are presented
in the table below by system organ class. These frequencies have been estimated on a per-patient basis
and are described using the following categories: very common (1/10); common (1/100 to <1/10);
and uncommon (1/1,000 to <1/100).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Frequency of Occurrence per Patient with ReF
acto or ReFacto AF
System Organ Class
Very common
(≥1/10)
Common
(≥1/100 to <1/10)
Uncommon
(1/1,000 to <1/100)
Blood and lymphatic
disorders
Factor VIII inhibitors -
PUPs
Factor VIII inhibitors -
PTPs
Metabolism and
nutrition disorders
Anorexia
Nervous system
disorders
Headache
Neuropathy, dizziness,
somnolence, dysgeusia
Cardiac disorders
Angina pectoris,
tachycardia, palpitations
Vascular disorders
Haemorrhage/haematoma Hypotension,
thrombophlebitis,
vasodilatation, flushing
Respiratory, thoracic
and mediastinal
disorders
Dyspnoea, cough
Gastrointestinal
disorders
Vomiting
Nausea
Abdominal pain,
diarrhoea
Skin and
subcutaneous tissue
disorders
Urticaria, pruritis, rash,
hyperhidrosis
Musculoskeletal,
connective tissue and
bone disorders
Arthralgia
Myalgia
General disorders and
administration site
conditions
Asthenia, pyrexia
Chills/feeling cold,
injection site
inflammation, injection
site reaction, injection
site pain
Investigations
Aspartate
aminotransferase
increased, alanine
aminotransferase
increased, blood
bilirubin increased,
blood creatine
phosphokinase increased
Surgical and medical
procedures
Vascular access
complication
Vascular access
complication
One event of cyst in an 11-year old patient and one event described as confusion in a 13-year old
patient have been reported as possibly related to ReFacto AF treatment.
17


































Safety of ReFacto AF was evaluated in previously treated children and adolescents (n=18, age 12-16
in a study and n=49, age 7-16 in a supporting study). Although a limited number of children have been
studied, there is a tendency for higher frequencies of adverse events in children aged 7-16 as compared
to adults. A clinical trial evaluating use of moroctocog alfa (AF-CC) in children less than 6 years of
age is on going.
The following adverse events have also been reported for ReFacto: paraesthesia, fatigue, blurred
vision, acne, gastritis, gastroenteritis, and pain.
Hypersensitivity or allergic reactions (which may include angioedema, burning and stinging at the
infusion site, chills, flushing, generalized urticaria, headache, hives, hypotension, lethargy, nausea,
restlessness, tachycardia, tightness of the chest, tingling, vomiting, wheezing) have been observed
infrequently for ReFacto, and may in some cases progress to severe anaphylaxis including shock (see
Trace amounts of hamster protein may be present ReFacto AF. Very rarely development of antibodies
to hamster protein has been observed, but there were no clinical sequelae. In a study of ReFacto,
twenty of 113 (18%) PTPs had an increase in anti-CHO antibody titre, without any apparent clinical
effect.
If any reaction takes place that is thought to be related to the administration of ReFacto AF, the rate of
infusion is to be decreased or the infusion stopped, as dictated by the response of the patient (see
No case of overdose has been reported.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antihaemorrhagics, blood coagulation factor VIII; ATC code: B02BD02.
ReFacto AF contains B-domain deleted recombinant coagulation factor VIII (moroctocog alfa). It is a
glycoprotein with an approximate molecular mass of 170,000 Da consisting of 1438 amino acids.
ReFacto AF has functional characteristics comparable to those of endogenous factor VIII. Factor VIII
activity is greatly reduced in patients with haemophilia A, and, therefore, replacement therapy is
necessary.
When infused into a haemophiliac patient, factor VIII binds to the von Willebrand factor present in the
patient’s circulation.
Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X
to activated factor X. Activated factor X converts prothrombin into thrombin. Thrombin then converts
fibrinogen into fibrin, and a clot is formed. Haemophilia A is a sex-linked hereditary disorder of blood
coagulation due to decreased levels of factor VIII:C and results in profuse bleeding into joints,
muscles or internal organs, either spontaneously or as a result of accidental or surgical trauma. By
replacement therapy, the plasma levels of factor VIII are increased, thereby enabling a temporary
correction of the factor deficiency and correction of the bleeding tendencies.
5.2 Pharmacokinetic properties
Pharmacokinetic properties of ReFacto, derived from a cross-over study of ReFacto and a plasma-
derived FVIII concentrate, using the chromogenic substrate assay (see section 4.2), in 18 previously
treated patients are listed in the table below.
18
Pharmacokinetic parameter estimates for ReFacto in previously treated patients with haemophilia A
PK parameter
Mean
SD
Median
AUC
t
(IU·h/ml)
19.9
4.9
19.9
t
1/2
(h)
14.8
5.6
12.7
CL (ml/h·kg)
2.4
0.75
2.3
MRT (h)
20.2
7.4
18.0
K-value
(IU/dl increase in FVIII:C per IU/kg FVIII given) 2.4 0.38 2.5
Abbreviations: AUC
t
= area under the plasma concentration-time curve from zero to the last measurable
concentration;
t
½
= half-life; CL = clearance; MRT = mean residence time; K-value = incremental recovery;
SD = standard deviation
In a study in which the potency of ReFacto AF, ReFacto and FVIII activity in patient plasma were
measured using the chromogenic substrate assay, ReFacto AF was shown to be bioequivalent to
ReFacto. The ratios of geometric least-square means of ReFacto AF-to-ReFacto were 100.6%, 99.5%
and 98.1% for K-value, AUC
t
and AUC
∞
(area under the plasma concentration curve from time zero
to infinity), respectively. The corresponding 90% confidence intervals about the ratios of ReFacto AF
to ReFacto geometric means were within the bioequivalence window of 80% to 125%, demonstratin
bioequivalence of ReFacto AF to ReFacto.
g
In a cross-over pharmacokinetic study, the pharmacokinetic parameters for ReFacto AF were
determined at baseline and followed-up in 25 previously treated patients (≥ 12 years) after repeated
administration of ReFacto AF for six months. The ratios of geometric least-square means of month
6-to-baseline pharmacokinetic were 107%, 100% and 104% for K-value, AUC
t
and AUC
,
respectively. The corresponding 90% confidence intervals about the ratios of month 6-to-baseline for
the above pharmacokinetic parameters were within the equivalence window of 80% to 125%. This
indicates no time-dependent changes in the pharmacokinetic properties of ReFacto AF.
In the same study, in which the drug potency of ReFacto AF and a full-length recombinant factor VIII
(FLrFVIII) comparator, and the FVIII activity measured in patient plasma samples were all
determined using the same one-stage clotting assay at a central laboratory, ReFacto AF was shown to
be pharmacokinetically equivalent to FLrFVIII in 30 previously treated patients ( 12 years) using the
standard bioequivalence approach.
In PUPs, pharmacokinetic parameters of ReFacto were evaluated using the chromogenic assay. These
patients (n=59; median age 10 ± 8.3 months) had a mean incremental recovery at Week 0 of
1.5 ± 0.6 IU/dl per IU/kg (range 0.2 to 2.8 IU/dl per IU/kg) which was lower than that obtained in
PTPs treated with ReFacto at Week 0 with a mean K-value of 2.4 ± 0.4 IU/dl per IU/kg (range 1.1 to
3.8 IU/dl per IU/kg). In the PUPs, the mean incremental recovery was stable over time (5 visits during
a 2-year period) and ranged from 1.5 to 1.8 IU/dl per IU/kg. Population pharmacokinetic modeling
using data from 44 PUPs led to a mean estimated half-life of 8.0 ± 2.2 hours.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity, and genotoxicity.
No investigations on carcinogenic potential or toxicity to reproduction have been conducted.
19










6.
6.1 List of excipients
Powder
Sucrose
Calcium chloride dihydrate
L-Histidine
Polysorbate 80
Sodium chloride
Solvent
Sodium chloride
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies this medicinal product must not be mixed with other medicinal
products, including other infusion solutions.
Only the provided infusion set is to be used, because treatment failure can occur as a consequence of
human-coagulation factor VIII adsorption to the internal surfaces of some infusion equipment.
6.3 Shelf life
Unopened powder vial
3 years.
After reconstitution
Chemical and physical in-use stability has been demonstrated for 3 hours at temperatures up to 25
o
C.
The product does not contain a preservative, and the reconstituted product should be used
immediately, or within 3 hours after reconstitution. Other in-use storage times and conditions are the
responsibility of the user.
6.4 Special precautions for storage
Store and transport refrigerated (2°C - 8°C). Do not freeze, in order to prevent damage to the pre-filled
syringe.
The product may be removed from refrigerated storage for one single period of maximum 3 months at
room temperature (up to 25°C). At the end of this period of room temperature storage, the product
must not be returned to refrigerated storage, but is to be used or discarded.
Keep the vial in the outer carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
6.5 Nature and contents of container
500 IU powder in a 10 ml vial (type 1 glass) with a stopper (butyl) and a flip-off seal (aluminum) and
4 ml of solvent in a pre-filled syringe (type 1 glass) with a plunger stopper (butyl), a tip-cap (butyl)
and a sterile vial adapter reconstitution device, a sterile infusion set, alcohol swabs, a plaster and a
gauze pad.
20
6.6
Special precautions for disposal and other handling
The vial of lyophilised product powder for injection must be reconstituted with the supplied solvent
[sodium chloride 9 mg/ml (0.9%) solution] from the pre-filled syringe using the sterile vial adapter
reconstitution device. The vial should be gently rotated until all of the powder is dissolved.
The product, when reconstituted, contains polysorbate-80, which is known to increase the rate of di-
(2-ethylhexyl)phthalate (DEHP) extraction from polyvinyl chloride (PVC). This is to be considered
during the preparation and administration of the product, including storage time elapsed in a PVC
container following reconstitution. It is important that the recommendations in section 6.3 be followed
closely.
After reconstitution, the solution is drawn back into the syringe. The solution will be clear or slightly
opalescent and colourless. The solution is to be discarded if visible particulate matter or discolouration
is observed.
Any unused product or waste material is to be disposed of in accordance with local requirements.
7.
Wyeth Europa Ltd.
Huntercombe Lane South
Taplow, Maidenhead
Berkshire, SL6 0PH
United Kingdom
8.
EU/1/99/103/002
9.
Date of first authorisation: 13 April 1999
Date of last renewal: 15 April 2009
Detailed information on this medicinal product is available on the website of the European Medicines
Agency (EMEA) http://www.emea.europa.eu/.
21
1.
ReFacto AF 1000 IU powder and solvent for solution for injection.
2.
Each vial contains nominally 1000 IU* moroctocog alfa**.
After reconstitution, each ml of solution contains approximately 250 IU moroctocog alfa.
* The potency (International Units) is determined using the European Pharmacopoeia chromogenic
assay. The specific activity of ReFacto AF is 7,600-13,800 IU/mg protein.
** Human coagulation factor VIII produced by recombinant DNA technology in Chinese hamster
ovary (CHO) cells. Moroctocog alfa is a glycoprotein with 1438 amino acids with a sequence that is
comparable to the 90 + 80 kDa form of factor VIII (i.e. B-domain deleted) and similar post-
translational modifications to those of the plasma-derived molecule.
The manufacturing process for ReFacto has been modified to eliminate any exogenous human- or
animal-derived protein in the cell culture process, purification, or final formulation; and at the same
time the invented name has been changed to ReFacto AF.
Excipients:
After reconstitution, 1.23 mmol (29 mg) sodium per vial.
For a full list of excipients, see section 6.1.
3.
Powder and solvent for solution for injection.
White to off-white cake/powder.
Clear, colourless solvent.
4.
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor VIII
deficiency).
ReFacto AF is appropriate for use in adults and children of all ages, including newborns.
ReFacto AF does not contain von Willebrand factor, and hence is not indicated in von Willebrand’s
disease.
Treatment should be initiated under the supervision of a physician experienced in the treatment of
haemophilia A.
Posology
22
The labelled potency of ReFacto AF is based on the European Pharmacopoeial chromogenic substrate
assay, in which the manufacturing potency standard has been calibrated to the WHO International
Standard using the chromogenic substrate assay. When monitoring patients' factor VIII activity levels
during treatment with ReFacto AF, use of the European Pharmacopoeial chromogenic substrate assay
is strongly recommended. The chromogenic assay yields results which are higher than those observed
with use of the one-stage clotting assay.
Typically, one-stage clotting assay results are 20-50% lower
than the chromogenic substrate assay results.
The ReFacto AF laboratory standard can be used to
correct for this discrepancy (see
section 5.2)
.
Another moroctocog alfa product approved for use outside Europe has a different potency assigned
using a manufacturing potency standard that has been calibrated to the WHO International Standard
using a one-stage clotting assay; this product is identified by the tradename XYNTHA. Due to the
difference in methods used to assign product potency of XYNTHA and ReFacto AF, 1 IU of the
XYNTHA product (one-stage assay calibrated) is approximately equivalent to 1.38 IU of the ReFacto
AF product (chromogenic assay calibrated). If a patient normally treated with XYNTHA is prescribed
ReFacto AF, the treating physician may consider adjustment of dosing recommendations based on
factor VIII recovery values.
Based on their current regimen, individuals with haemophilia A should be advised to bring an
adequate supply of factor VIII product for anticipated treatment when travelling. Patients should be
advised to consult with their healthcare provider prior to travel.
The dosage and duration of the substitution therapy depend on the severity of the factor VIII
deficiency, on the location and extent of bleeding, and on the patient’s clinical condition. Doses
administered should be titrated to the patient's clinical response. In the presence of an inhibitor, higher
doses or appropriate specific treatment may be required.
The number of units of factor VIII administered is expressed in International Units (IUs), which are
related to the current WHO standard for factor VIII products. Factor VIII activity in plasma is
expressed either as a percentage (relative to normal human plasma) or in International Units (relative
to an International Standard for factor VIII in plasma). One International Unit (IU) of factor VIII
activity is equivalent to the quantity of factor VIII in one ml of normal human plasma. The calculation
of the required dosage of factor VIII is based upon the empirical finding that 1 International Unit (IU)
of factor VIII per kg body weight raises the plasma factor VIII activity by 2 IU/dl. The required
dosage is determined using the following formula:
Required units (IU) = body weight (kg) x desired factor VIII rise (% or IU/dl) x 0.5 (IU/kg per IU/dl),
where 0.5 IU/kg per IU/dl represents the reciprocal of the incremental recovery generally observed
following infusions of factor VIII.
The amount to be administered and the frequency of administration should always be oriented to the
clinical effectiveness in the individual case.
In the case of the following haemorrhagic events, the factor VIII activity should not fall below the
given plasma levels (in % of normal or in IU/dl) in the corresponding period. The following table can
be used to guide dosing in bleeding episodes and surgery:
23
Degree of haemorrhage/
Type of surgical procedure
Factor VIII level
required (% or IU/dl)
Frequency of doses (hours)/
Duration of therapy (days)
Haemorrhage
Early haemarthrosis, muscle
bleeding or oral bleeding
20-40
Repeat every 12-24 hours. At least 1
day until the bleeding episode as
indicated by pain is resolved or healing
is achieved.
More extensive haemarthrosis,
muscle bleeding or haematoma
30-60
Repeat infusion every 12-24 hours for
3-4 days or more until pain and acute
disability are resolved.
Life-threatening haemorrhages
60-100
Repeat infusion every 8-24 hours until
threat is resolved.
Surgery
Minor,
including tooth extraction
30-60
Every 24 hours, at least 1 day, until
healing is achieved.
Major
80-100
(pre- and
post-operative)
Repeat infusion every 8-24 hours until
adequate wound healing, then therapy
for at least another 7 days to maintain a
factor VIII activity of 30% to 60%
(IU/dl).
During the course of treatment, appropriate determination of factor VIII levels is advised to guide the
dose to be administered and the frequency of repeated infusions. In the case of major surgical
interventions in particular, precise monitoring of the substitution therapy by means of coagulation
analysis (plasma factor VIII activity) is indispensable. Individual patients may vary in their response
to factor VIII, achieving different levels of
in vivo
recovery and demonstrating different half-lives.
For long-term prophylaxis against bleeding in patients with severe haemophilia A, the usual doses are
20 to 40 IU of factor VIII per kg body weight at intervals of 2 to 3 days. In some cases, especially in
younger patients, shorter dosage intervals or higher doses may be necessary.
Patients using factor VIII replacement therapy are to be monitored for the development of factor VIII
inhibitors. If expected factor VIII activity plasma levels are not attained, or if bleeding is not
controlled with an appropriate dose, an assay should be performed to determine if factor VIII
inhibitors are present. Data from clinical trials indicated that if inhibitors are present at levels less than
10 Bethesda Units (BUs), administration of additional antihaemophilic factor may neutralise the
inhibitors. In patients with levels of inhibitor above 10 BU, factor VIII therapy may not be effective
and other therapeutic options should be considered. Management of such patients should be directed
by physicians with experience in the care of patients with haemophilia (see
section 4.4)
.
Special populations
Renal or hepatic impairment
Dosage adjustment for patients with renal or hepatic impairment has not been studied in clinical trials.
Paediatric patients
Safety and efficacy studies with ReFacto have been performed both in previously treated children and
adolescents (n=31, ages 8-18 years) and in previously untreated neonates, infants and children (n=101,
ages < 1-52 months).
24


The need for an increased dose relative to that used for adults and older children should be anticipated
when treating younger children with ReFacto AF. In a study of ReFacto in children less than 6 years
of age, pharmacokinetic analysis revealed half-life and recovery less than that observed in older
children and adults (see section 5.2). During the clinical trials, children less than 6 years of age on a
prophylaxis regimen used an average dose of 50 IU/kg of ReFacto and experienced an average of
6.1 bleeding episodes per year. Older children and adults on a prophylaxis regimen used an average
dose of 27 IU/kg and experienced an average of 10 bleeding episodes per year. In a clinical trial
setting the mean dose per infusion of ReFacto for bleeding episodes in children less than 6 years of
age was higher than the mean dose administered to older children and adults (51.3 IU/kg and
29.3 IU/kg, respectively).
Method of administration
ReFacto AF is administered by intravenous injection over several minutes after reconstitution of the
lyophilised powder for injection with sodium chloride 9 mg/ml (0.9%) solution for injection
(provided). The rate of administration should be determined by the patient’s comfort level.
Appropriate training is recommended for non-healthcare professionals administering the product.
In the interest of patients, it is recommended that every time ReFacto AF is administered, the name
and batch number of the product should be recorded.
For reconstitution instructions prior to administration, see section 6.6.
Hypersensitivity to the active substance or to any of the excipients.
Hypersensitivity to hamster proteins.
As with any intravenous protein product, allergic-type hypersensitivity reactions are possible. The
product contains traces of hamster proteins. Patients should be informed of the early signs of
hypersensitivity reactions (including hives, generalised urticaria, tightness of the chest, wheezing,
hypotension) and anaphylaxis. If allergic or anaphylactic reactions occur, administration of ReFacto
AF is to be discontinued immediately, and an appropriate treatment must be initiated. In case of shock,
the current medical standards for treatment of shock are to be observed. Patients are to be advised to
discontinue use of the product and contact their physician or seek immediate emergency care,
depending on the type and severity of the reaction, if any of these symptoms occur.
The formation of neutralising antibodies (inhibitors) to factor VIII is a known complication in the
management of individuals with haemophilia A. These inhibitors are usually IgG immunoglobulins
directed against the factor VIII procoagulant activity, which are quantified in Bethesda Units (BUs)
per ml of plasma using the Nijmegen modification of the Bethesda assay. The risk of developing
inhibitors is correlated to the exposure to factor VIII, this risk being highest within the first
20 exposure days. Inhibitors have been observed in previously treated patients receiving factor VIII
products, including ReFacto AF. Cases of recurrence of inhibitors (low titre) have been observed after
switching from one recombinant factor VIII product to another in previously treated patients with
more than 100 exposure days who have a history of inhibitor development. Patients treated with
recombinant coagulation factor VIII should be carefully monitored for the development of inhibitors
by appropriate clinical observations and laboratory tests (see also
section 4.8)
.
Reports of lack of effect, mainly in prophylaxis patients, have been received in the clinical trials and in
the post-marketing setting for ReFacto. The reported lack of effect with ReFacto has been described as
bleeding into target joints, bleeding into new joints or a subjective feeling by the patient of new onset
25
bleeding. When prescribing ReFacto AF it is important to individually titrate and monitor each
patient’s factor level in order to ensure an adequate therapeutic response.
In the interest of patient safety, it is recommended that every time ReFacto AF is administered, the
name on the carton and batch number of the product are recorded. Patients can affix one of the peel-
off labels found on the vial to document the batch number in their diary or for reporting any side
effects.
After reconstitution this medicinal product contains 1.23 mmol (29 mg) sodium per vial, to be taken
into consideration by patients on a controlled sodium diet.
No interaction studies have been performed.
Animal reproduction studies have not been conducted with factor VIII. Because of the rare occurrence
of haemophilia A in women, experience regarding the use of factor VIII during pregnancy and breast-
feeding is not available. Therefore, factor VIII should be used during pregnancy and lactation only if
clearly indicated.
No studies on the effects on the ability to drive and use machines have been performed.
Factor VIII inhibition
The occurrence of neutralising antibodies (inhibitors) to factor VIII is well known in the treatment of
patients with haemophilia A. As with all coagulation factor VIII products, patients are to be monitored
for the development of inhibitors that are to be titrated in Bethesda Units (BUs) using the Nijmegen
modification of the Bethesda assay.If such inhibitors occur, the condition may manifest itself as an
insufficient clinical response. In such cases, it is recommended that a specialised haemophilia centre
be contacted.
In a clinical study with ReFacto AF in previously treated patients (PTPs), the incidence of factor VIII
inhibitors was the primary safety endpoint. Two clinically silent, low-titre, transient inhibitors were
observed in 94 patients with a median exposure of 76 exposure days (ED, range 1-92), corresponding
to 2.2% of the 89 patients with at least 50 ED. In a supporting study of ReFacto AF, 1
de novo
and 2
recurrent inhibitors (all low-titre, central laboratory determination) were observed in 110 patients;
median exposure of 58 ED (range 5-140) and 98 patients had at least 50 ED to ReFacto AF.
Ninety-eight (98) of the original 110 patients continued treatment in a second supportive study and
had subsequent extended exposure to ReFacto AF with a median of 169 additional ED (range 9-425).
One (1) additional low-titre
de novo
inhibitor was observed. The frequency of inhibitors observed in
these studies is within the expected range.
In a clinical study with ReFacto PTPs, 1 inhibitor was observed in 113 patients. Also, there have been
spontaneous post-marketing reports of high-titre inhibitors involving previously treated patients.
There are no clinical data on previously untreated patients (PUPs) with ReFacto AF. However, clinical
trials are planned in previously untreated patients (PUPs) with ReFacto AF. In a clinical trial, 32 out of
101 (32%) previously untreated patients (PUPs) treated with ReFacto developed inhibitors: 16 out of
101 (16%) with a titre > 5 BU and 16 out of 101 (16%) with a titre ≤ 5 BU. The median number of
exposure days up to inhibitor development in these patients was 12 (range 3-49). Of the 16 patients
with high titres, 15 received immune tolerance (IT) treatment. Of the 16 patients with low titres, IT
26
treatment was started in 10. IT had an efficacy of 73% for patients with high titres and 90% for those
with low titres. For all 101 treated PUPs, regardless of inhibitor development, the median number of
exposure days is 197 (range 1-1299).
Adverse reactions based on experience from clinical trials with ReFacto or ReFacto AF are presented
in the table below by system organ class. These frequencies have been estimated on a per-patient basis
and are described using the following categories: very common (1/10); common (1/100 to <1/10);
and uncommon (1/1,000 to <1/100).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
System organ class
Frequency of Occurrence per Patient with
ReFacto or ReFacto AF
Very common
(≥1/10)
Common
(≥1/100 to <1/10)
Uncommon
(1/1,000 to <1/100)
Blood and lymphatic
disorders
Factor VIII inhibitors -
PUPs
Factor VIII inhibitors -
PTPs
Metabolism and
nutrition disorders
Anorexia
Nervous system
disorders
Headache
Neuropathy, dizziness,
somnolence, dysgeusia
Cardiac disorders
Angina pectoris,
tachycardia, palpitations
Vascular disorders
Haemorrhage/haematoma Hypotension,
thrombophlebitis,
vasodilatation, flushing
Respiratory, thoracic
and mediastinal
disorders
Dyspnoea, cough
Gastrointestinal
disorders
Vomiting
Nausea
Abdominal pain,
diarrhoea
Skin and
subcutaneous tissue
disorders
Urticaria , pruritis, rash,
hyperhidrosis
Musculoskeletal,
connective tissue and
bone disorders
Arthralgia
Myalgia
General disorders and
administration site
conditions
Asthenia, pyrexia
Chills/feeling cold,
injection site
inflammation, injection
site reaction, injection
site pain
Investigations
Aspartate
aminotransferase
increased, alanine
aminotransferase
increased, blood
bilirubin increased,
blood creatine
phosphokinase increased
Surgical and medical
procedures
Vascular access
complication
One event of cyst in an 11-year old patient and one event described as confusion in a 13-year old
patient have been reported as possibly related to ReFacto AF treatment.
27

































Safety of ReFacto AF was evaluated in previously treated children and adolescents (n=18, age 12-16
in a study and n=49, age 7-16 in a supporting study). Although a limited number of children have been
studied, there is a tendency for higher frequencies of adverse events in children aged 7-16 as compared
to adults.A clinical trial evaluating use of moroctocog alfa (AF-CC) in children less than 6 years of
age is on going.
The following adverse events have also been reported for ReFacto: paraesthesia, fatigue, blurred
vision, acne, gastritis, gastroenteritis, and pain.
Hypersensitivity or allergic reactions (which may include angioedema, burning and stinging at the
infusion site, chills, flushing, generalized urticaria, headache, hives, hypotension, lethargy, nausea,
restlessness, tachycardia, tightness of the chest, tingling, vomiting, wheezing) have been observed
infrequently for ReFacto, and may in some cases progress to severe anaphylaxis including shock (see
Trace amounts of hamster protein may be present in ReFacto AF. Very rarely, development of
antibodies to hamster protein has been observed, but there were no clinical sequelae. In a study of
ReFacto, twenty of 113 (18%) PTPs had an increase in anti-CHO antibody titre, without any apparent
clinical effect.
If any reaction takes place that is thought to be related to the administration of ReFacto AF, the rate of
infusion is to be decreased or the infusion stopped, as dictated by the response of the patient (see
No case of overdose has been reported.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antihaemorrhagics, blood coagulation factor VIII; ATC code: B02BD02.
ReFacto AF contains B-domain deleted recombinant coagulation factor VIII, (moroctocog alfa). It is a
glycoprotein with an approximate molecular mass of 170,000 Da consisting of 1438 amino acids.
ReFacto AF has functional characteristics comparable to those of endogenous factor VIII. Factor VIII
activity is greatly reduced in patients with haemophilia A, and, therefore, replacement therapy is
necessary.
When infused into a haemophiliac patient, factor VIII binds to the von Willebrand factor present in the
patient’s circulation.
Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X
to activated factor X. Activated factor X converts prothrombin into thrombin. Thrombin then converts
fibrinogen into fibrin, and a clot is formed. Haemophilia A is a sex-linked hereditary disorder of blood
coagulation due to decreased levels of factor VIII:C and results in profuse bleeding into joints,
muscles or internal organs, either spontaneously or as a result of accidental or surgical trauma. By
replacement therapy the plasma levels of factor VIII are increased, thereby enabling a temporary
correction of the factor deficiency and correction of the bleeding tendencies.
5.2 Pharmacokinetic properties
Pharmacokinetic properties of ReFacto, derived from a cross-over study of ReFacto and a plasma-
derived FVIII concentrate, using the chromogenic substrate assay (see section 4.2), in 18 previously
treated patients are listed in the table below.
28
Pharmacokinetic parameter estimates for ReFacto in previously treated patients with haemophilia A
PK parameter
Mean
SD
Median
AUC
t
(IU·h/ml)
19.9
4.9
19.9
t
1/2
(h)
14.8
5.6
12.7
CL (ml/h·kg)
2.4
0.75
2.3
MRT (h)
20.2
7.4
18.0
K-value
(IU/dl increase in FVIII:C per IU/kg FVIII given) 2.4 0.38 2.5
Abbreviations: AUC
t
= area under the plasma concentration-time curve from zero to the last measurable
concentration;
t
½
= half-life; CL = clearance; MRT = mean residence time; K-value = incremental recovery;
SD = standard deviation
In a study in which the potency of ReFacto AF, ReFacto and FVIII activity in patient plasma were
measured using the chromogenic substrate assay, ReFacto AF was shown to be bioequivalent to
ReFacto. The ratios of geometric least-square means of ReFacto AF-to-ReFacto were 100.6%, 99.5%
and 98.1% for K-value, AUC
t
and AUC
∞
(area under the plasma concentration curve from time zero
infinity), respectively. The corresponding 90% confidence intervals about the ratios of ReFacto AF to
ReFacto geometric means were within the bioequivalence window of 80% to 125%, demonstrating
bioequivalence of ReFacto AF to ReFacto.
In a cross-over pharmacokinetic study, the pharmacokinetic parameters for ReFacto AF were
determined at baseline and followed-up in 25 previously treated patients (≥ 12 years) after repeated
administration of ReFacto AF for six months. The ratios of geometric least-square means of month 6-
to-baseline pharmacokinetic were 107%, 100% and 104% for K-value, AUC
t
and AUC
,
respectively.
The corresponding 90% confidence intervals about the ratios of month 6-to-baseline for the above
pharmacokinetic parameters were within the equivalence window of 80% to 125%. This indicates no
time-dependent changes in the pharmacokinetic properties of ReFacto AF.
In the same study, in which the drug potency of ReFacto AF and a full-length recombinant factor VIII
(FLrFVIII) comparator, and the FVIII activity measured in patient plasma samples were all
determined using the same one-stage clotting assay at a central laboratory, ReFacto AF was shown to
be pharmacokinetically equivalent to FLrFVIII in 30 previously treated patients ( 12 years) using the
standard bioequivalence approach.
In PUPs, pharmacokinetic parameters of ReFacto were evaluated using the chromogenic assay. These
patients (n=59; median age 10 ± 8.3 months) had a mean incremental recovery at Week 0 of
1.5 ± 0.6 IU/dl per IU/kg (range 0.2 to 2.8 IU/dl per IU/kg) which was lower than that obtained in
PTPs treated with ReFacto at Week 0 with a mean K-value of 2.4 ± 0.4 IU/dl per IU/kg (range 1.1 to
3.8 IU/dl per IU/kg). In the PUPs, the mean incremental recovery was stable over time (5 visits during
a 2-year period) and ranged from 1.5 to 1.8 IU/dl per IU/kg. Population pharmacokinetic modeling
using data from 44 PUPs led to a mean estimated half-life of 8.0 ± 2.2 hours.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity, and genotoxicity.
No investigations on carcinogenic potential or toxicity to reproduction have been conducted.
29










6.
6.1 List of excipients
Powder
Sucrose
Calcium chloride dihydrate
L-Histidine
Polysorbate 80
Sodium chloride
Solvent
Sodium chloride
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products, including other infusion solutions.
Only the provided infusion set is to be used, because treatment failure can occur as a consequence of
human-coagulation factor VIII adsorption to the internal surfaces of some infusion equipment.
6.3 Shelf life
Unopened powder vial
3 years.
After reconstitution
Chemical and physical in-use stability has been demonstrated for 3 hours at temperatures up to 25
o
C.
The product does not contain a preservative, and the reconstituted product should be used
immediately, or within 3 hours after reconstitution. Other in-use storage times and conditions are the
responsibility of the user.
6.4 Special precautions for storage
Store and transport refrigerated (2°C - 8°C). Do not freeze, in order to prevent damage to the pre-filled
syringe.
The product may be removed from refrigerated storage for one single period of maximum 3 months at
room temperature (up to 25°C). At the end of this period of room temperature storage, the product
must not be returned to refrigerated storage, but is to be used or discarded.
Keep the vial in the outer carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
30
6.5 Nature and contents of container
1000 IU powder in a 10 ml vial (type 1 glass) with a stopper (butyl) and a flip-off seal (aluminum) and
4 ml of solvent in a pre-filled syringe (type 1 glass) with a plunger stopper (butyl), a tip-cap (butyl)
and a sterile vial adapter reconstitution device, a sterile infusion set, alcohol swabs, a plaster and a
gauze pad.
6.6
Special precautions for disposal and other handling
The vial of lyophilised product powder for injection must be reconstituted with the supplied solvent
[sodium chloride 9 mg/ml (0.9%) solution] from the pre-filled syringe using the sterile vial adapter
reconstitution device. The vial should be gently rotated until all of the powder is dissolved.
The product, when reconstituted, contains polysorbate-80, which is known to increase the rate of di-
(2-ethylhexyl)phthalate (DEHP) extraction from polyvinyl chloride (PVC). This is to be considered
during the preparation and administration of the product, including storage time elapsed in a PVC
container following reconstitution. It is important that the recommendations in section 6.3 be followed
closely.
After reconstitution, the solution is drawn back into the syringe. The solution will be clear or slightly
opalescent and colourless. The solution is to be discarded if visible particulate matter or discolouration
is observed.
Any unused product or waste material is to be disposed of in accordance with local requirements.
7.
Wyeth Europa Ltd.
Huntercombe Lane South
Taplow, Maidenhead
Berkshire, SL6 0PH
United Kingdom
8.
EU/1/99/103/003
9.
Date of first authorisation: 13 April 1999
Date of last renewal: 15 April 2009
Detailed information on this medicinal product is available on the website of the European Medicines
Agency (EMEA) http://www.emea.europa.eu/.
31
1.
ReFacto AF 2000 IU powder and solvent for solution for injection.
2.
Each vial contains nominally 2000 IU* moroctocog alfa**.
After reconstitution, each ml of solution contains approximately 500 IU moroctocog alfa.
* The potency (International Units) is determined using the European Pharmacopoeia chromogenic
assay. The specific activity of ReFacto AF is 7,600-13,800 IU/mg protein.
** Human coagulation factor VIII produced by recombinant DNA technology in Chinese hamster
ovary (CHO) cells. Moroctocog alfa is a glycoprotein with 1438 amino acids with a sequence that is
comparable to the 90 + 80 kDa form of factor VIII (i.e. B-domain deleted) and similar post-
translational modifications to those of the plasma-derived molecule.
The manufacturing process for ReFacto has been modified to eliminate any exogenous human- or
animal-derived protein in the cell culture process, purification, or final formulation; and at the same
time the invented name has been changed to ReFacto AF.
Excipients:
After reconstitution, 1.23 mmol (29 mg) sodium per vial.
For a full list of excipients, see section 6.1.
3.
Powder and solvent for solution for injection.
White to off-white cake/powder.
Clear, colourless solvent.
4.
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor VIII
deficiency).
ReFacto AF is appropriate for use in adults and children of all ages, including newborns.
ReFacto AF does not contain von Willebrand factor, and hence is not indicated in von Willebrand’s
disease.
Treatment should be initiated under the supervision of a physician experienced in the treatment of
haemophilia A.
Posology
32
The labelled potency of ReFacto AF is based on the European Pharmacopoeial chromogenic substrate
assay, in which the manufacturing potency standard has been calibrated to the WHO International
Standard using the chromogenic substrate assay. When monitoring patients' factor VIII activity levels
during treatment with ReFacto AF, use of the European Pharmacopoeial chromogenic substrate assay
is strongly recommended. The chromogenic assay yields results which are higher than those observed
with use of the one-stage clotting assay.
Typically, one-stage clotting assay results are 20-50% lower
than the chromogenic substrate assay results.
The ReFacto AF laboratory standard can be used to
correct for this discrepancy (see
section 5.2)
.
Another moroctocog alfa product approved for use outside Europe has a different potency assigned
using a manufacturing potency standard that has been calibrated to the WHO International Standard
using a one-stage clotting assay; this product is identified by the tradename XYNTHA. Due to the
difference in methods used to assign product potency of XYNTHA and ReFacto AF, 1 IU of the
XYNTHA product (one-stage assay calibrated) is approximately equivalent to 1.38 IU of the ReFacto
AF product (chromogenic assay calibrated). If a patient normally treated with XYNTHA is prescribed
ReFacto AF, the treating physician may consider adjustment of dosing recommendations based on
factor VIII recovery values.
Based on their current regimen, individuals with haemophilia A should be advised to bring an
adequate supply of factor VIII product for anticipated treatment when travelling. Patients should be
advised to consult with their healthcare provider prior to travel.
The dosage and duration of the substitution therapy depend on the severity of the factor VIII
deficiency, on the location and extent of bleeding, and on the patient’s clinical condition. Doses
administered should be titrated to the patient's clinical response. In the presence of an inhibitor, higher
doses or appropriate specific treatment may be required.
The number of units of factor VIII administered is expressed in International Units (IUs), which are
related to the current WHO standard for factor VIII products. Factor VIII activity in plasma is
expressed either as a percentage (relative to normal human plasma) or in International Units (relative
to an International Standard for factor VIII in plasma). One International Unit (IU) of factor VIII
activity is equivalent to the quantity of factor VIII in one ml of normal human plasma. The calculation
of the required dosage of factor VIII is based upon the empirical finding that 1 International Unit (IU)
of factor VIII per kg body weight raises the plasma factor VIII activity by 2 IU/dl. The required
dosage is determined using the following formula:
Required units (IU) = body weight (kg) x desired factor VIII rise (% or IU/dl) x 0.5 (IU/kg per IU/dl),
where 0.5 IU/kg per IU/dl represents the reciprocal of the incremental recovery generally observed
following infusions of factor VIII.
The amount to be administered and the frequency of administration should always be oriented to the
clinical effectiveness in the individual case.
In the case of the following haemorrhagic events, the factor VIII activity should not fall below the
given plasma levels (in % of normal or in IU/dl) in the corresponding period. The following table can
be used to guide dosing in bleeding episodes and surgery:
33
Degree of haemorrhage/
Type of surgical procedure
Factor VIII level
required (% or IU/dl)
Frequency of doses (hours)/
Duration of therapy (days)
Haemorrhage
Early haemarthrosis, muscle
bleeding or oral bleeding
20-40
Repeat every 12-24 hours. At least 1
day until the bleeding episode as
indicated by pain is resolved or healing
is achieved.
More extensive haemarthrosis,
muscle bleeding or haematoma
30-60
Repeat infusion every 12-24 hours for
3-4 days or more until pain and acute
disability are resolved.
Life-threatening haemorrhages
60-100
Repeat infusion every 8-24 hours until
threat is resolved.
Surgery
Minor,
including tooth extraction
30-60
Every 24 hours, at least 1 day, until
healing is achieved.
Major
80-100
(pre- and
post-operative)
Repeat infusion every 8-24 hours until
adequate wound healing, then therapy
for at least another 7 days to maintain a
factor VIII activity of 30% to 60%
(IU/dl).
During the course of treatment, appropriate determination of factor VIII levels is advised to guide the
dose to be administered and the frequency of repeated infusions. In the case of major surgical
interventions in particular, precise monitoring of the substitution therapy by means of coagulation
analysis (plasma factor VIII activity) is indispensable. Individual patients may vary in their response
to factor VIII, achieving different levels of
in vivo
recovery and demonstrating different half-lives.
For long-term prophylaxis against bleeding in patients with severe haemophilia A, the usual doses are
20 to 40 IU of factor VIII per kg body weight at intervals of 2 to 3 days. In some cases, especially in
younger patients, shorter dosage intervals or higher doses may be necessary.
Patients using factor VIII replacement therapy are to be monitored for the development of factor VIII
inhibitors. If expected factor VIII activity plasma levels are not attained, or if bleeding is not
controlled with an appropriate dose, an assay should be performed to determine if factor VIII
inhibitors are present. Data from clinical trials indicated that if inhibitors are present at levels less than
10 Bethesda Units (BUs), administration of additional antihaemophilic factor may neutralise the
inhibitors. In patients with levels of inhibitor above 10 BU, factor VIII therapy may not be effective
and other therapeutic options should be considered. Management of such patients should be directed
by physicians with experience in the care of patients with haemophilia (see
section 4.4)
.
Special populations
Renal or hepatic impairment
Dosage adjustment for patients with renal or hepatic impairment has not been studied in clinical trials.
Paediatric patients
Safety and efficacy studies with ReFacto have been performed both in previously treated children and
adolescents (n=31, ages 8-18 years) and in previously untreated neonates, infants and children (n=101,
ages < 1-52 months).
34


The need for an increased dose relative to that used for adults and older children should be anticipated
when treating younger children with ReFacto AF. In a study of ReFacto in children less than 6 years
of age, pharmacokinetic analysis revealed half-life and recovery less than that observed in older
children and adults (see section 5.2). During the clinical trials, children less than 6 years of age on a
prophylaxis regimen used an average dose of 50 IU/kg of ReFacto and experienced an average of
6.1 bleeding episodes per year. Older children and adults on a prophylaxis regimen used an average
dose of 27 IU/kg and experienced an average of 10 bleeding episodes per year. In a clinical trial
setting the mean dose per infusion of ReFacto for bleeding episodes in children less than 6 years of
age was higher than the mean dose administered to older children and adults (51.3 IU/kg and
29.3 IU/kg, respectively).
Method of administration
ReFacto AF is administered by intravenous injection over several minutes after reconstitution of the
lyophilised powder for injection with sodium chloride 9 mg/ml (0.9%) solution for injection
(provided). The rate of administration should be determined by the patient’s comfort level.
Appropriate training is recommended for non-healthcare professionals administering the product.
In the interest of patients, it is recommended that every time ReFacto AF is administered, the name
and batch number of the product should be recorded.
Hypersensitivity to the active substance or to any of the excipients.
Hypersensitivity to hamster proteins.
As with any intravenous protein product, allergic-type hypersensitivity reactions are possible. The
product contains traces of hamster proteins. Patients should be informed of the early signs of
hypersensitivity reactions (including hives, generalised urticaria, tightness of the chest, wheezing,
hypotension) and anaphylaxis. If allergic or anaphylactic reactions occur, administration of ReFacto
AF is to be discontinued immediately, and an appropriate treatment must be initiated. In case of shock,
the current medical standards for treatment of shock are to be observed. Patients are to be advised to
discontinue use of the product and contact their physician or seek immediate emergency care,
depending on the type and severity of the reaction, if any of these symptoms occur.
The formation of neutralising antibodies (inhibitors) to factor VIII is a known complication in the
management of individuals with haemophilia A. These inhibitors are usually IgG immunoglobulins
directed against the factor VIII procoagulant activity, which are quantified in Bethesda Units (BUs)
per ml of plasma using the Nijmegen modification of the Bethesda assay. The risk of developing
inhibitors is correlated to the exposure to factor VIII, this risk being highest within the first
20 exposure days. Inhibitors have been observed in previously treated patients receiving factor VIII
products, including ReFacto AF. Cases of recurrence of inhibitors (low titre) have been observed after
switching from one recombinant factor VIII product to another in previously treated patients with
more than 100 exposure days who have a history of inhibitor development. Patients treated with
recombinant coagulation factor VIII should be carefully monitored for the development of inhibitors
by appropriate clinical observations and laboratory tests (see also
section 4.8)
.
Reports of lack of effect, mainly in prophylaxis patients, have been received in the clinical trials and in
the post-marketing setting for ReFacto. The reported lack of effect with ReFacto has been described as
bleeding into target joints, bleeding into new joints or a subjective feeling by the patient of new onset
bleeding. When prescribing ReFacto AF it is important to individually titrate and monitor each
patient’s factor level in order to ensure an adequate therapeutic response.
35
In the interest of patient safety, it is recommended that every time ReFacto AF is administered, the
name on the carton and batch number of the product are recorded. Patients can affix one of the peel-
off labels found on the vial to document the batch number in their diary or for reporting any side
effects.
After reconstitution this medicinal product contains 1.23 mmol (29 mg) sodium per vial, to be taken
into consideration by patients on a controlled sodium diet.
No interaction studies have been performed.
Animal reproduction studies have not been conducted with factor VIII. Because of the rare occurrence
of haemophilia A in women, experience regarding the use of factor VIII during pregnancy and breast-
feeding is not available. Therefore, factor VIII should be used during pregnancy and lactation only if
clearly indicated.
No studies on the effects on the ability to drive and use machines have been performed.
Factor VIII inhibition
The occurrence of neutralising antibodies (inhibitors) to factor VIII is well known in the treatment of
patients with haemophilia A. As with all coagulation factor VIII products, patients are to be monitored
for the development of inhibitors that are to be titrated in Bethesda Units (BUs) using the Nijmegen
modification of the Bethesda assay. If such inhibitors occur, the condition may manifest itself as an
insufficient clinical response. In such cases, it is recommended that a specialised haemophilia centre
be contacted.
In a clinical study with ReFacto AF in previously treated patients (PTPs), the incidence of factor VIII
inhibitors was the primary safety endpoint. Two clinically silent, low-titre, transient inhibitors were
observed in 94 patients with a median exposure of 76 exposure days (ED, range 1-92), corresponding
to 2.2% of the 89 patients with at least 50 ED. In a supporting study of ReFacto AF, 1
de novo
and 2
recurrent inhibitors (all low-titre, central laboratory determination) were observed in 110 patients;
median exposure of 58 ED (range 5-140) and 98 patients had at least 50 ED to ReFacto AF.
Ninety-eight (98) of the original 110 patients continued treatment in a second supportive study and
had subsequent extended exposure to ReFacto AF with a median of 169 additional ED (range 9-425).
One (1) additional low-titre
de novo
inhibitor was observed. The frequency of inhibitors observed in
these studies is within the expected range.
In a clinical study with ReFacto in PTPs, 1 inhibitor was observed in 113 patients. Also, there have
been spontaneous post-marketing reports of high-titre inhibitors involving previously treated patients.
There are no clinical data on previously untreated patients (PUPs) with ReFacto AF. However, clinical
trials are planned in previously untreated patients (PUPs) with ReFacto AF. In a clinical trial, 32 out of
101 (32%) previously untreated patients (PUPs) treated with ReFacto developed inhibitors: 16 out of
101 (16%) with a titre > 5 BU and 16 out of 101 (16%) with a titre ≤ 5 BU. The median number of
exposure days up to inhibitor development in these patients was 12 (range 3-49). Of the 16 patients
with high titres, 15 received immune tolerance (IT) treatment. Of the 16 patients with low titres, IT
treatment was started in 10. IT had an efficacy of 73% for patients with high titres and 90% for those
with low titres. For all 101 treated PUPs, regardless of inhibitor development, the median number of
exposure days is 197 (range 1-1299).
36
Adverse reactions based on experience from clinical trials with ReFacto or ReFacto AF are presented
in the table below by system organ class. These frequencies have been estimated on a per-patient basis
and are described using the following categories: very common (1/10); common (1/100 to <1/10);
and uncommon (1/1,000 to <1/100).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Frequency of Occurrence per Patient with ReF
acto or ReFacto AF
System Organ Class
Very common
(≥1/10)
Common
(≥1/100 to <1/10)
Uncommon
(1/1,000 to <1/100)
Blood and lymphatic
disorders
Factor VIII inhibitors -
PUPs
Factor VIII inhibitors -
PTPs
Metabolism and
nutrition disorders
Anorexia
Nervous system
disorders
Headache
Neuropathy, dizziness,
somnolence, dysgeusia
Cardiac disorders
Angina pectoris,
tachycardia, palpitations
Vascular disorders
Haemorrhage/haematoma Hypotension,
thrombophlebitis,
vasodilatation, flushing
Respiratory, thoracic
and mediastinal
disorders
Dyspnoea, cough
Gastrointestinal
disorders
Vomiting
Nausea
Abdominal pain,
diarrhoea
Skin and
subcutaneous tissue
disorders
Urticaria, pruritis, rash,
hyperhidrosis
Musculoskeletal,
connective tissue and
bone disorders
Arthralgia
Myalgia
General disorders and
administration site
conditions
Asthenia, pyrexia
Chills/feeling cold,
injection site
inflammation, injection
site reaction, injection
site pain
Investigations
Aspartate
aminotransferase
increased, alanine
aminotransferase
increased, blood
bilirubin increased,
blood creatine
phosphokinase increased
Surgical and medical
procedures
Vascular access
complication
One event of cyst in an 11-year old patient and one event described as confusion in a 13-year old
patient have been reported as possibly related to ReFacto AF treatment.
Safety of ReFacto AF was evaluated in previously treated children and adolescents (n=18, age 12-16
in a study and n=49, age 7-16 in a supporting study). Although a limited number of children have been
studied, there is a tendency for higher frequencies of adverse events in children aged 7-16 as compared
37


































to adults. A clinical trial evaluating use of moroctocog alfa (AF-CC) in children less than 6 years of
age is on going.
The following adverse events have also been reported for ReFacto: paraesthesia, fatigue, blurred
vision, acne, gastritis, gastroenteritis, and pain.
Hypersensitivity or allergic reactions (which may include angioedema, burning and stinging at the
infusion site, chills, flushing, generalized urticaria, headache, hives, hypotension, lethargy, nausea,
restlessness, tachycardia, tightness of the chest, tingling, vomiting, wheezing) have been observed
infrequently for ReFacto, and may in some cases progress to severe anaphylaxis including shock (see
Trace amounts of hamster protein may be present in ReFacto AF. Very rarely, development of
antibodies to hamster protein has been observed, but there were no clinical sequelae. In a study of
ReFacto, twenty of 113 (18%) PTPs had an increase in anti-CHO antibody titre, without any apparent
clinical effect.
If any reaction takes place that is thought to be related to the administration of ReFacto AF, the rate of
infusion is to be decreased or the infusion stopped, as dictated by the response of the patient (see
No case of overdose has been reported.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antihaemorrhagics, blood coagulation factor VIII; ATC code: B02BD02.
ReFacto AF contains B-domain deleted recombinant coagulation factor VIII, (moroctocog alfa). It is a
glycoprotein with an approximate molecular mass of 170,000 Da consisting of 1438 amino acids.
ReFacto AF has functional characteristics comparable to those of endogenous factor VIII. Factor VIII
activity is greatly reduced in patients with haemophilia A, and, therefore, replacement therapy is
necessary.
When infused into a haemophiliac patient, factor VIII binds to the von Willebrand factor present in the
patient’s circulation.
Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X
to activated factor X. Activated factor X converts prothrombin into thrombin. Thrombin then converts
fibrinogen into fibrin, and a clot is formed. Haemophilia A is a sex-linked hereditary disorder of blood
coagulation due to decreased levels of factor VIII:C and results in profuse bleeding into joints,
muscles or internal organs, either spontaneously or as a result of accidental or surgical trauma. By
replacement therapy, the plasma levels of factor VIII are increased, thereby enabling a temporary
correction of the factor deficiency and correction of the bleeding tendencies.
5.2 Pharmacokinetic properties
Pharmacokinetic properties of ReFacto, derived from a cross-over study of ReFacto and a plasma-
derived FVIII concentrate, using the chromogenic substrate assay (see section 4.2), in 18 previously
treated patients are listed in the table below.
38
Pharmacokinetic parameter estimates for ReFacto in previously treated patients with haemophilia A
PK parameter
Mean
SD
Median
AUC
t
(IU·h/ml)
19.9
4.9
19.9
t
1/2
(h)
14.8
5.6
12.7
CL (ml/h·kg)
2.4
0.75
2.3
MRT (h)
20.2
7.4
18.0
K-value
(IU/dl increase in FVIII:C per IU/kg FVIII given) 2.4 0.38 2.5
Abbreviations: AUC
t
= area under the plasma concentration-time curve from zero to the last measurable
concentration;
t
½
= half-life; CL = clearance; MRT = mean residence time; K-value = incremental recovery;
SD = standard deviation
In a study in which the potency of ReFacto AF, ReFacto and FVIII activity in patient plasma were
measured using the chromogenic substrate assay, ReFacto AF was shown to be bioequivalent to
ReFacto. The ratios of geometric least-square means of ReFacto AF-to-ReFacto were 100.6%, 99.5%
and 98.1% for K-value, AUC
t
and AUC
∞
(area under the plasma concentration curve from time zero
to infinity), respectively. The corresponding 90% confidence intervals about the ratios of ReFacto AF
to ReFacto geometric means were within the bioequivalence window of 80% to 125%, demonstratin
bioequivalence of ReFacto AF to ReFacto.
g
In a cross-over pharmacokinetic study, the pharmacokinetic parameters for ReFacto AF were
determined at baseline and followed-up in 25 previously treated patients (≥ 12 years) after repeated
administration of ReFacto AF for six months. The ratios of geometric least-square means of month 6-
to-baseline pharmacokinetic were 107%, 100% and 104% for K-value, AUC
t
and AUC
,
respectively.
The corresponding 90% confidence intervals about the ratios of month 6-to-baseline for the above
pharmacokinetic parameters were within the equivalence window of 80% to 125%. This indicates no
time-dependent changes in the pharmacokinetic properties of ReFacto AF.
In the same study, in which the drug potency of ReFacto AF and a full-length recombinant factor VIII
(FLrFVIII) comparator, and the FVIII activity measured in patient plasma samples were all
determined using the same one-stage clotting assay at a central laboratory, ReFacto AF was shown to
be pharmacokinetically equivalent to FLrFVIII in 30 previously treated patients (12 years) using the
standard bioequivalence approach.
In PUPs, pharmacokinetic parameters of ReFacto were evaluated using the chromogenic assay. These
patients (n=59; median age 10 ± 8.3 months) had a mean incremental recovery at Week 0 of
1.5 ± 0.6 IU/dl per IU/kg (range 0.2 to 2.8 IU/dl per IU/kg) which was lower than that obtained in
PTPs treated with ReFacto at Week 0 with a mean K-value of 2.4
±
0.4 IU/dl per IU/kg (range 1.1 to
3.8 IU/dl per IU/kg). In the PUPs, the mean incremental recovery was stable over time (5 visits during
a 2-year period) and ranged from 1.5 to 1.8 IU/dl per IU/kg. Population pharmacokinetic modeling
using data from 44 PUPs led to a mean estimated half-life of 8.0 ± 2.2 hours.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity, and genotoxicity.
No investigations on carcinogenic potential or toxicity to reproduction have been conducted.
39










6.
6.1 List of excipients
Powder
Sucrose
Calcium chloride dihydrate
L-Histidine
Polysorbate 80
Sodium chloride
Solvent
Sodium chloride
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products, including other infusion solutions.
Only the provided infusion set is to be used, because treatment failure can occur as a consequence of
human-coagulation factor VIII adsorption to the internal surfaces of some infusion equipment.
6.3 Shelf life
Unopened powder vial
3 years.
After reconstitution
Chemical and physical in-use stability has been demonstrated for 3 hours at temperatures up to 25
o
C.
The product does not contain a preservative, and the reconstituted product should be used
immediately, or within 3 hours after reconstitution. Other in-use storage times and conditions are the
responsibility of the user.
6.4 Special precautions for storage
Store and transport refrigerated (2°C - 8°C). Do not freeze, in order to prevent damage to the pre-filled
syringe.
The product may be removed from refrigerated storage for one single period of maximum 3 months at
room temperature (up to 25°C). At the end of this period of room temperature storage, the product
must not be returned to refrigerated storage, but is to be used or discarded.
Keep the vial in the outer carton in order to protect from light.
For storage conditions of the reconstituted medicinal product, see section 6.3.
40
6.5 Nature and contents of container
2000 IU powder in a 10 ml vial (type 1 glass) with a stopper (butyl) and a flip-off seal (aluminum) and
4 ml of solvent in a pre-filled syringe (type 1 glass) with a plunger stopper (butyl), a tip-cap (butyl)
and a sterile vial adapter reconstitution device, a sterile infusion set, alcohol swabs, a plaster and a
gauze pad.
6.6 Special precautions for disposal and other handling
The vial of lyophilised product powder for injection must be reconstituted with the supplied solvent
[sodium chloride 9 mg/ml (0.9%) solution] from the pre-filled syringe using the sterile vial adapter
reconstitution device. The vial should be gently rotated until all of the powder is dissolved.
The product, when reconstituted, contains polysorbate-80, which is known to increase the rate of di-
(2-ethylhexyl)phthalate (DEHP) extraction from polyvinyl chloride (PVC). This is to be considered
during the preparation and administration of the product, including storage time elapsed in a PVC
container following reconstitution. It is important that the recommendations in section 6.3 be followed
closely.
After reconstitution, the solution is drawn back into the syringe. The solution will be clear or slightly
opalescent and colourless. The solution is to be discarded if visible particulate matter or discolouration
is observed.
Any unused product or waste material is to be disposed of in accordance with local requirements.
7.
Wyeth Europa Ltd.
Huntercombe Lane South
Taplow, Maidenhead
Berkshire, SL6 0PH
United Kingdom
8.
EU/1/99/103/004
9.
Date of first authorisation: 13 April 1999
Date of last renewal: 15 April 2009
Detailed information on this medicinal product is available on the website of the European Medicines
Agency (EMEA) http://www.emea.europa.eu/.
41
ANNEX II
A.
MANUFACTURER OF THE BIOLOGICAL ACTIVE
SUBSTANCE AND MANUFACTURING AUTHORISATION
HOLDER RESPONSIBLE FOR BATCH RELEASE
B.
CONDITIONS OF THE MARKETING AUTHORISATION
42
A. MANUFACTURER OF THE BIOLOGICAL ACTIVE SUBSTANCE AND
MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturer of the biological active substance
Biovitrum AB (publ)
Strandbergsgatan 49
SE-11276 Stockholm
Sweden
Name and address of the manufacturers responsible for batch release
Wyeth Farma S.A
Autovia del Norte A-1 Km 23
28700 San Sebastian de los Reyes
Madrid
Spain
B. CONDITIONS OF THE MARKETING AUTHORISATION
Medicinal product subject to restricted medical prescription (see Annex I: Summary of Product
Characteristics, section
4.2)
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND EFFECTIVE
USE OF THE MEDICINAL PRODUCT
The Marketing Authorisation Holder (MAH) should ensure that, at launch, all Healthcare
Professionals who are expected to prescribe/use ReFacto AF, all laboratories that are expected to
monitor patients receiving ReFacto AF and all EU Haemophiliac patients associations are provided
with Educational packs.
The educational pack should contain the following
Summary of Product Characteristics and Patient Information Leaflet for ReFacto AF
The educational materials.
The educational pack for Healthcare Professionals should include the educational materials to be
provided by prescribers to patients before they receive RefactoAF.
The educational materials for Healthcare professionals, patients and haemophiliac patients
associations should include the following key elements:
The main differences between ReFacto AF and ReFacto
Changes highlighted in the Summary of Product Characteristics and Patient
Information Leaflet for ReFacto AF.
The visual points of differentiation in packaging for ReFacto AF versus ReFacto.
Specific instructions regarding the proper identification, dosing and monitoring of
ReFacto AF.
43
Desvio Algete Km 1
That after switching to ReFacto AF, patients should remain on ReFacto AF and not
switch back to ReFacto.
The potential risks for medication errors in using different assays or laboratory
standard for patient monitoring Information that the chromogenic substrate assay is
strongly recommended to be used by laboratories when monitoring patients receiving
ReFacto AF and that typically one stage clotting assay results are 20-50% lower than
the chromogenic substrate assay results.
The existence of another moroctocog alfa containing product for use outside Europe
with a different potency assigned using a one stage clotting essay and the need for
patients to take an adequate supply of their ReFacto AF for anticipated treatment
while traveling. Advice for health care professionals on possible need to adjust
dosages for patients normally treated outside Europe with Xyntha.
The importance to report suspected adverse reactions (including inhibitor occurrence)
detailing the name and the batch number of the product used. The importance to
report medication errors, and their causes and consequences.
Instructions on record keeping with recommendation to record, the name and batch
number of the product received, using the peel-off labels provided on the vial.
Additional messages regarding the transition plan for replacement of ReFacto with
ReFacto AF in the Member State(s).
The educational programme for laboratories should inform about the following key elements:
The main differences between ReFacto AF and ReFacto.
Specific instructions regarding the proper monitoring of ReFacto AF.
The potential risks for medication errors in using different assays or laboratory
standard for patient monitoring.
Strong recommendation to use the chromogenic substrate assay when monitoring
patients. Information that typically one stage clotting assay results are 20-50% lower
than the chromogenic substrate assay results.
The purpose of the laboratory standard. The differences in the new laboratory standard
for ReFacto AF as compared to the laboratory standard for ReFacto and instructions
on when to change to using the ReFacto AF laboratory standard.
Additional messages regarding the transition plan for replacement of ReFacto with
ReFacto AF in the Member State(s)
The Marketing Authorisation Holder shall agree the educational material with the national
competent authorities in all the Member States prior to the launch of the product
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, as described in version 3.0 presented in
Module 1.8.1. of the Marketing Authorisation Application, is in place and functioning before and
whilst the product is on the market.
44
Risk Management Plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in
the Pharmacovigilance Plan, as agreed in version 4.7 of the Risk Management Plan (RMP) presented
in Module 1.8.2. of the Marketing Authorisation Application and any subsequent updates of the RMP
agreed by the CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, the
updated RMP should be submitted at the same time as the next Periodic Safety Update Report
(PSUR).
In addition, an updated RMP should be submitted
When new information is received that may impact on the current Safety Specification,
Pharmacovigilance Plan or risk minimisation activities
Within 60 days of an important (pharmacovigilance or risk minimisation) milestone being
reached
At the request of the EMEA
PSUR cycle
The MAH is required to submit PSURs at 6-monthly intervals for the first two years. Then PSURs
should be submited once a year for the following 3 years.
45
ANNEX III
LABELLING AND PACKAGE LEAFLET
46
A. LABELLING
47
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
REFACTO AF OUTER CARTON
1.
ReFacto AF 250 IU powder and solvent for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
One vial contains 250 IU moroctocog alfa. After reconstitution, each ml of solution contains
approximately 62.5 IU moroctocog alfa.
3.
LIST OF EXCIPIENTS
Sucrose,
calcium chloride dihydrate,
L-histidine,
polysorbate 80,
sodium chloride
. Read the package leaflet before use.
4.
Powder and solvent for solution for injection
Each pack contains:
1 vial with 250 IU moroctocog alfa
1 pre-filled syringe with 4 ml sterile Solvent
1 Vial Adapter
1 Sterile Infusion Set
2 Alcohol Swabs
1 Plaster
1 Gauze
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Read the
package leaflet
before use.
Intravenous use, after reconstitution.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
48























7.
OTHER SPECIAL WARNING(S), IF NECESSARY
Do not use after expiry date
8.
EXPIRY DATE
EXP
Use immediately or within 3 hours of reconstitution.
9.
SPECIAL STORAGE CONDITIONS
Store and transport at 2C – 8C.
Do not freeze
Keep the vial in the original package in order to protect from light.
ReFacto AF can be stored at room temperature (up to 25
o
C) for a single period up to 3 months. The
product may not be returned to refrigerated storage after storage at room temperature.
Date removed from the refrigerator:
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Discard any remaining reconstituted solution
Wyeth Europa Ltd
Huntercombe Lane South
Taplow, Maidenhead
Berkshire, SL6 0PH
United Kingdom
EU/1/99/103/001
13. MANUFACTURER’S BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
49



























Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
ReFacto AF 250
50





MINIMUM PARTICULARS TO APPEAR ON BLISTERS OR STRIPS
REFACTO AF TRAY LID
1.
ReFacto AF 250 IU powder and solvent for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
2.
Wyeth Europa Ltd
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
Intravenous use
Read the package leaflet before use
Keep this container in the outer carton
Store in a refrigerator
51
















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
REFACTO AF VIAL LABEL
1.
ReFacto AF 250 IU powder for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
IV use
2.
METHOD OF ADMINISTRATION
Read the package leaflet before use.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
6.
OTHER
Store in a refrigerator
52






















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
REFACTO AF SOLVENT PRE-FILLED SYRINGE LABEL
1.
Solvent for ReFacto AF
2. METHOD OF ADMINISTRATION
IV use, after reconstitution.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
Contains 4 ml of sodium chloride 9 mg/ml (0.9%) solution for injection
6.
OTHER
Store in a refrigerator
53
















PARTICULARS TO APPEAR ON THE OUTER PACKAGING
REFACTO AF OUTER CARTON
1.
ReFacto AF 500 IU powder and solvent for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
One vial contains 500 IU moroctocog alfa. After reconstitution, each ml of solution contains
approximately 125 IU moroctocog alfa.
3.
LIST OF EXCIPIENTS
Sucrose,
calcium chloride dihydrate,
L-histidine,
polysorbate 80,
sodium chloride
. Read the package leaflet before use.
4.
Powder and solvent for solution for injection
Each pack contains:
1 vial with 500 IU moroctocog alfa
1 pre-filled syringe with 4 ml sterile Solvent
1 Vial Adapter
1 Sterile Infusion Set
2 Alcohol Swabs
1 Plaster
1 Gauze
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Read the
package leaflet
before use.
Intravenous use, after reconstitution.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
54























7.
OTHER SPECIAL WARNING(S), IF NECESSARY
Do not use after expiry date
8.
EXPIRY DATE
EXP
Use immediately or within 3 hours of reconstitution.
9.
SPECIAL STORAGE CONDITIONS
Store and transport at 2C – 8C.
Do not freeze
Keep the vial in the original package in order to protect from light.
ReFacto AF can be stored at room temperature (up to 25
o
C) for a single period up to 3 months. The
product may not be returned to refrigerated storage after storage at room temperature.
Date removed from the refrigerator:
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Discard any remaining reconstituted solution
Wyeth Europa Ltd
Huntercombe Lane South
Taplow, Maidenhead
Berkshire, SL6 0PH
United Kingdom
EU/1/99/103/002
13. MANUFACTURER’S BATCH NUMBER
Lot
55























14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
ReFacto AF 500
56







MINIMUM PARTICULARS TO APPEAR ON BLISTERS OR STRIPS
REFACTO AF TRAY LID
1.
ReFacto AF 500 IU powder and solvent for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
2.
Wyeth Europa Ltd
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
Intravenous use
Read the package leaflet before use
Keep this container in the outer carton
Store in a refrigerator
57
















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
REFACTO AF VIAL LABEL
1.
ReFacto AF 500 IU powder for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
IV use
2.
METHOD OF ADMINISTRATION
Read the package leaflet before use.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
6.
OTHER
Store in a refrigerator
58




















PARTICULARS TO APPEAR ON THE OUTER PACKAGING
REFACTO AF OUTER CARTON
1.
ReFacto AF 1000 IU powder and solvent for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
One vial contains 1000 IU moroctocog alfa. After reconstitution, each ml of solution contains
approximately 250 IU moroctocog alfa.
3.
LIST OF EXCIPIENTS
Sucrose,
calcium chloride dihydrate,
L-histidine,
polysorbate 80,
sodium chloride
. Read the package leaflet before use.
4.
Powder and solvent for solution for injection
Each pack contains:
1 vial with 1000 IU moroctocog alfa
1 pre-filled syringe with 4 ml sterile Solvent
1 Vial Adapter
1 Sterile Infusion Set
2 Alcohol Swabs
1 Plaster
1 Gauze
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Read the
package leaflet
before use.
Intravenous use, after reconstitution.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
59























7.
OTHER SPECIAL WARNING(S), IF NECESSARY
Do not use after expiry date
8.
EXPIRY DATE
EXP
Use immediately or within 3 hours of reconstitution.
9.
SPECIAL STORAGE CONDITIONS
Store and transport at 2C – 8C.
Do not freeze
Keep the vial in the original package in order to protect from light.
ReFacto AF can be stored at room temperature (up to 25
o
C) for a single period up to 3 months. The
product may not be returned to refrigerated storage after storage at room temperature.
Date removed from the refrigerator:
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Discard any remaining reconstituted solution
Wyeth Europa Ltd
Huntercombe Lane South
Taplow, Maidenhead
Berkshire, SL6 0PH
United Kingdom
EU/1/99/103/003
13. MANUFACTURER’S BATCH NUMBER
Lot
60























14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
ReFacto AF 1000
61







MINIMUM PARTICULARS TO APPEAR ON BLISTERS OR STRIPS
REFACTO AF TRAY LID
1.
ReFacto AF 1000 IU powder and solvent for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
2.
Wyeth Europa Ltd
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
Intravenous use
Read the package leaflet before use
Keep this container in the outer carton
Store in a refrigerator
62
















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
REFACTO AF VIAL LABEL
1.
ReFacto AF 1000 IU powder for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
IV use
2.
METHOD OF ADMINISTRATION
Read the package leaflet before use.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
6.
OTHER
Store in a refrigerator
63


















PARTICULARS TO APPEAR ON THE OUTER PACKAGING
REFACTO AF OUTER CARTON
1.
ReFacto AF 2000 IU powder and solvent for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
One vial contains 2000 IU moroctocog alfa. After reconstitution, each ml of solution contains
approximately 500 IU moroctocog alfa.
3.
LIST OF EXCIPIENTS
Sucrose,
calcium chloride dihydrate,
L-histidine,
polysorbate 80,
sodium chloride
. Read the package leaflet before use.
4.
Powder and solvent for solution for injection
Each pack contains:
1 vial with 2000 IU moroctocog alfa
1 pre-filled syringe with 4 ml sterile Solvent
1 Vial Adapter
1 Sterile Infusion Set
2 Alcohol Swabs
1 Plaster
1 Gauze
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Read the
package leaflet
before use.
Intravenous use, after reconstitution.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
64























7.
OTHER SPECIAL WARNING(S), IF NECESSARY
Do not use after expiry date
8.
EXPIRY DATE
EXP
Use immediately or within 3 hours of reconstitution.
9.
SPECIAL STORAGE CONDITIONS
Store and transport at 2C – 8C.
Do not freeze
Keep the vial in the original package in order to protect from light.
ReFacto AF can be stored at room temperature (up to 25
o
C) for a single period up to 3 months. The
product may not be returned to refrigerated storage after storage at room temperature.
Date removed from the refrigerator:
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Discard any remaining reconstituted solution
Wyeth Europa Ltd
Huntercombe Lane South
Taplow, Maidenhead
Berkshire, SL6 0PH
United Kingdom
EU/1/99/103/004
13. MANUFACTURER’S BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
65



























Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
ReFacto AF 2000
66





MINIMUM PARTICULARS TO APPEAR BLISTERS OR STRIPS
REFACTO AF TRAY LID
1.
ReFacto AF 2000 IU powder and solvent for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
2.
Wyeth Europa Ltd
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
Intravenous use
Read the package leaflet before use
Keep this container in the outer carton
Store in a refrigerator
67
















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
REFACTO AF VIAL LABEL
1.
ReFacto AF 2000 IU powder for solution for injection
Moroctocog alfa
(recombinant human coagulation factor VIII)
IV use
2.
METHOD OF ADMINISTRATION
Read the package leaflet before use.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
6.
OTHER
Store in a refrigerator
68




















PACKAGE LEAFLET: INFORMATION FOR THE USER
ReFacto AF 250 IU powder and solvent for solution for injection
ReFacto AF 500 IU powder and solvent for solution for injection
ReFacto AF 1000 IU powder and solvent for solution for injection
ReFacto AF 2000 IU powder and solvent for solution for injection
Moroctocog alfa (recombinant human coagulation factor VIII)
Read all of this leaflet carefully before you start taking this medicine.
-
Keep this leaflet. You may need to read it again.
-
If you have further questions, please ask your doctor or your pharmacist.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
-
If you are concerned about any side effects or if you notice any side effects not listed in this
leaflet, please tell your doctor or pharmacist.
In this leaflet:
1.
What ReFacto AF is and what it is used for
2.
Before you use ReFacto AF
4.
Possible side effects
5
How to store ReFacto AF
6.
Further information
1.
WHAT REFACTO AF IS AND WHAT IT IS USED FOR
ReFacto AF contains the active substance moroctocog alfa, human coagulation factor VIII produced
by recombinant DNA technology. Factor VIII is necessary for the blood to form clots and stop
bleedings. In patients with haemophilia A (inborn factor VIII deficiency), it is missing or not working
properly.
ReFacto AF is used for the treatment and prevention of bleeding (prophylaxis) in adults and children
of all ages (including newborns) with haemophilia A.
ReFacto AF does not contain von Willebrand factor, and hence is not indicated in von Willebrand’s
disease.
2.
BEFORE YOU USE REFACTO AF
Do not use ReFacto AF
-
if you are allergic (hypersensitive) to moroctocog alfa or any of the other ingredients of ReFacto
AF (listed in
section 6
).
-
if you are hypersensitive to hamster proteins.
If you are unsure about this, ask your doctor.
Take special care with ReFacto AF
70
3.
How to use ReFacto AF
-
if you experience allergic reactions. Some of the signs of allergic (hypersensitivity) reactions are
difficulty in breathing, shortness of breath, swelling, hives, itching, tightness of the chest,
wheezing, and low blood pressure. Anaphylaxis is a severe allergic reaction that can cause
difficulty in swallowing and/or breathing, red or swollen face and/or hands. If any of these signs
occur, stop the infusion immediately and contact a doctor or seek immediate emergency care. In
case of severe allergic reactions, alternative therapy must be considered.
-
if your bleeding does not stop as expected and contact your doctor or seek immediate
emergency care.
-
if bleeding is not adequately controlled with the usual dose. Patients receiving factor VIII
products may sometimes develop antibodies to factor VIII (also known as factor VIII
inhibitors), which may prevent the factor VIII product from working properly. While being
treated with ReFacto AF, you should be monitored for the development of factor VIII inhibitors.
-
if you are elderly. Your doctor may change the dose.
Taking other medicines
Please tell your doctor or pharmacist if you are taking, or have recently taken, any other medicines,
including medicines obtained without a prescription.
Pregnancy and breast-feeding
Tell your doctor if you are pregnant or breast-feeding.
Your doctor will decide if ReFacto AF may be used during pregnancy and breast-feeding.
Driving and using machines
No studies on the effects on the ability to drive and use machines have been performed.
Important information about some of the ingredients of ReFacto AF
This medicinal product contains 1.23 mmol (or 29 mg) sodium per vial of reconstituted powder.Inform
your doctor if you are on a controlled sodium diet.
3.
HOW TO USE REFACTO AF
Always take ReFacto AF exactly as your doctor has instructed you. You should check with your
doctor or pharmacist if you are not sure.
Your doctor will decide the dose of ReFacto AF you will receive. This dose and duration will depend
upon your individual needs for replacement factor VIII therapy.
During your treatment, your doctor may decide to change the dose of ReFacto AF you receive.
Consult with your health care provider before you travel. You should bring enough of your factor VIII
product for anticipated treatment when travelling.
It is recommended that every time you use ReFacto AF, you record the name on the carton and batch
number of the product. You can use one of the peel-off labels found on the vial to document the batch
number in your diary or for reporting any side effects.
Reconstitution and administration
71
The procedures below are provided as guidelines for the reconstitution and administration of ReFacto
AF. Patients should follow the specific reconstitution and administration procedures provided by their
doctors.
Use only the pre-filled syringe provided in the box for reconstitution. Other sterile disposable syringes
may be used for administration.
ReFacto AF is administered by intravenous (IV) injection after reconstitution of the lyophilised
powder for injection with the supplied solvent [sodium chloride 9 mg/ml (0.9%) solution] syringe.
ReFacto AF should not be mixed with other infusion solutions.
Always wash your hands before performing the following reconstitution and administration
procedures. Aseptic technique (meaning clean and germ-free) should be used during the reconstitution
procedure.
Reconstitution:
1.
Allow the vial of lyophilised ReFacto AF and the pre-filled solvent syringe to reach room
temperature.
2.
Remove the plastic flip-top cap from the ReFacto AF vial to expose the central portion of the
rubber stopper.
3.
Wipe the top of the vial with the alcohol swab provided, or use another antiseptic solution and
allow to dry. After cleaning, do not touch the rubber stopper with your hand or allow it to touch
any surface.
4.
Peel back the lid from the clear plastic vial adapter package. Do not remove the adapter from the
package.
5.
Place the vial on a flat surface. While holding the adapter package, place the vial adapter over
the vial. Press down firmly on the package until the adapter snaps into place on top of the vial,
with the adapter spike penetrating the vial stopper.
72

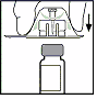
6.
Lift the package away from the adapter and discard the package.
7.
Attach the plunger rod to the solvent syringe by inserting the rod into the opening in the syringe
stopper and pushing and turning the rod firmly until it is securely seated in the stopper.
8.
Break off the tamper-resistant plastic tip cap from the solvent syringe by snapping the
perforation of the cap. This is done by bending the cap up and down until the perforation is
broken. Do not touch the inside of the cap or the syringe tip. The cap may need to be replaced
(if not administering reconstituted ReFacto AF immediately), so set it aside by placing it on its
top.
9.
Place the vial on a flat surface. Connect the solvent syringe to the vial adapter by inserting the
tip of the syringe into the adapter opening while firmly pushing and turning the syringe
clockwise until the connection is secured.
73


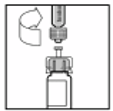
10. Slowly depress the plunger rod to inject all the solvent into the ReFacto AF vial.
11. With the syringe still connected to the adapter,
gently
rotate the vial until the powder is
dissolved.
12. The final solution must be inspected visually for particulate matter before administration. The
solution will appear clear to slightly opalescent and colourless.
Note: If you use more than one vial of ReFacto AF per infusion, each vial should be
reconstituted as per the previous instructions. The solvent syringe should be removed, leaving
the vial adapter in place, and a single large luer lock syringe may be used to draw back the
reconstituted contents of each of the individual vials.
13. Ensuring that the syringe plunger rod is still fully depressed, invert the vial. Slowly draw back
all the solution through the vial adapter into the syringe.
14.
Detach the syringe from the vial adapter by gently pulling and turning the syringe counter-
clockwise. Discard the vial with the adapter attached.
Note: If the solution is not to be used immediately, the syringe cap is to be carefully replaced.
Do not touch the syringe tip or the inside of the cap.
ReFacto AF must be used within 3 hours of reconstitution. The reconstituted solution may be stored at
room temperature prior to administration.
Administration (Intravenous Injection):
ReFacto AF should be administered using the infusion set provided in this kit and the pre-filled
solvent syringe provided or a single sterile disposable plastic luer lock syringe.
1.
Attach the syringe to the luer end of the infusion set tubing.
74


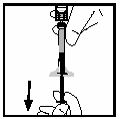
2.
Apply a tourniquet and prepare the injection site by wiping the skin well with an alcohol swab
provided in the kit.
3.
Insert the needle on the infusion set tubing into the vein as instructed by your doctor, and
remove the tourniquet. Remove any air in the infusion set tubing by drawing back on the
syringe. The reconstituted product is to be injected intravenously over several minutes. Your
doctor may change your recommended infusion rate to make the infusion more comfortable.
Please dispose of all unused solution, the empty vial(s) and the used needles and syringes in an
appropriate container for throwing away of medical waste as these materials may hurt others if not
disposed of properly.
If you take more ReFacto AF than you should
Check with your doctor or pharmacist.
If you stop taking ReFacto AF
Do not stop using ReFacto AF without consulting your doctor.
If you have any further questions regarding this product, ask your doctor or pharmacist.
4.
POSSIBLE SIDE EFFECTS
Like all medicines, ReFacto AF can cause side effects, although not everybody gets them.
Inhibitor development
Patients with haemophilia A may develop neutralising antibodies (inhibitors) to factor VIII. If such
inhibitors occur, a sign may be an increase in the amount of ReFacto AF typically required to treat a
bleed and/or continued bleeding after a treatment. In such cases, it is recommended that a specialised
haemophilia centre be contacted. Your doctor may want to monitor you for inhibitor development.
Development of inhibitors occurred in approximately 2% of patients receiving ReFacto AF in a
research study.
If you experience a significant increase in your usage of ReFacto AF in order to control a bleed, please
contact your doctor immediately.
Side effects may occur with certain frequencies, which are defined as follows:
very common: affects more than 1 user in 10
common: affects 1 to 10 users in 100
uncommon: affects 1 to 10 users in 1,000
rare: affects 1 to 10 users in 10,000
75


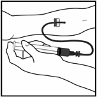
very rare: affects less than 1 user in 10,000
not known: frequency cannot be estimated from the available data.
Very common side effects
Vomiting
Inhibitor development for patients who have never been previously treated with factor VIII
products
Common side effects
Bleeding
Inhibitor development for patients who have been previously treated with factor VIII products
headache, nausea
joint pain, muscular pain
fatigue, fever
venous access catheter complications
Uncommon side effects
Severe allergic reaction, dizziness, light-headedness, hives, itching, rash
chest pain, shortness of breath, rapid heart beat
diarrhoea, loss of appetite, stomach pain
altered taste
chills, sweating, muscle weakness
sleepiness
coughing
injection site reactions (including burning and stinging at the infusion site), discomfort and
swelling at the intravenous site
slight increase in heart enzymes
increased liver enzymes, increased bilirubin
The following adverse reactions have also been reported: numbness, blurred vision, acne,
gastroenteritis, and pain.
Hypersensitivity/allergic reactions
If
severe, sudden allergic reactions
(anaphylactic) occur, the injection
must be stopped
immediately
. You must
contact your doctor immediately
if you have any of the following early
symptoms of allergic (hypersensitivity) reactions:
rash, hives, wheals, generalised itching
swelling of lips and tongue
difficulty in breathing, wheezing, tightness in the chest
general feeling of being unwell
dizziness and loss of consciousness
Severe symptoms, including difficulty in breathing and (nearly) fainting, require prompt emergency
treatment.
If you are concerned about any side effects, or if you notice any side effects not listed in this leaflet,
please tell your doctor or pharmacist.
Patients with haemophilia A may develop neutralising antibodies (inhibitors) to factor VIII. If such
inhibitors occur, a sign may be an increase in the amount of ReFacto AF typically required to treat a
bleed and/or continued bleeding after a treatment. In such cases, it is recommended that a specialised
haemophilia centre be contacted. Your doctor may want to monitor you for inhibitor development.
76
5.
HOW TO STORE REFACTO AF
Keep out of the reach and sight of children.
Do not use after the expiry date stated on the outer carton and vial label after EXP. The expiry date
refers to the last day of that month.
Store and transport refrigerated (2C – 8C). Do not freeze, in order to prevent damage to the
pre-filled solvent syringe.
For your convenience, the product can be removed from such storage for one single period of
maximum 3 months at room temperature (up to 25°C). At the end of this room temperature storage
period, the product must not be put back in the refrigerator, but must be used or discarded. Record on
the outer carton the date ReFacto AF is removed from the refrigerator and set at room temperature (up
to 25
o
C). Keep the vial in the outer carton in order to protect from light.
Use the reconstituted solution within 3 hours of reconstitution.
The solution will be clear to slightly opalescent and colourless. Do not use ReFacto AF if you notice
that it is cloudy or contains visible particles.
Medicines must not be disposed of via wastewater or household waste. Ask your pharmacist how to
dispose of medicines no longer required. These measures will help to protect the environment.
6.
FURTHER INFORMATION
What ReFacto AF contains
-
The active substance is moroctocog alfa (recombinant coagulation factor VIII). Each vial of
ReFacto AF contains nominally 250, 500, 1000, or 2000 IU of moroctocog alfa.
-
The other ingredients are sucrose, calcium chloride dihydrate, L-histidine, polysorbate 80 and
sodium chloride. A solvent [sodium chloride 9 mg/ml (0.9%) solution for injection] is also
supplied for reconstitution.
-
After reconstitution with the supplied solvent [sodium chloride 9 mg/ml (0.9%) solution], each
vial contains 62.5, 125, 250, or 500 IU, respectively (based on the strength of moroctocog alfa,
i.e., 250, 500, 1000, or 2000 IU), of moroctocog alfa per 1 ml of the prepared solution for
injection.
What ReFacto AF looks like and contents of the pack
ReFacto AF is provided as a powder for injection in a glass vial and a solvent is provided in a pre-
filled syringe.
The contents of the pack are:
-
one pre-filled syringe of solvent, 4 ml sterile sodium chloride 9 mg/ml (0.9 %) solution for
injection for reconstitution, with one plunger rod
-
one sterile vial adapter reconstitution device
-
two alcohol swabs
77
-
one vial of moroctocog alfa 250, 500, 1000, or 2000 IU powder
-
one sterile infusion set
-
one gauze pad
Marketing Authorisation Holder
Wyeth Europa Ltd.
Huntercombe Lane South
Taplow, Maidenhead
Berkshire, SL6 0PH
United Kingdom
Manufacturers
Wyeth Farma S.A.
Autovia del Norte A-1 Km 23
Desvio Algete Km 1
28700 San Sebastian de los Reyes
Madrid
Spain
For any information about this medicinal product, please contact the local representative of the
Marketing Authorisation Holder.
België/Belgique/Belgien
Luxembourg/Luxemburg
Pfizer S.A./ N.V.
Tél/Tel: +32 (0)2 554 62 11
Magyarország
Pfizer Kft
Tel :+36 1 488 3700
България/Eesti/Latvija/Lietuva/
Slovenija
Wyeth Whitehall Export GmbH
Teл./Tel/Tãlr: +43 1 89 1140
Malta
Vivian Corporation Ltd.
Tel: +356 21 344616
Česká republika
Pfizer s.r.o.
Tel: +420-283-004-111
Nederland
Wyeth Pharmaceuticals B.V.
Tel: +31 23 567 2567
Danmark
Pfizer ApS
Tlf: +45 44 201 100
Norge
Pfizer AS
Tlf: +47 67 526 100
Deutschland
Pfizer Pharma GmbH
Tel: +49 (0)30 550055-51000
Österreich
Pfizer Corporation Austria Ges.m.b.H.
Tel: +43 (0)1 521 15-0
Ελλάδα
Pfizer Hellas A.E.
Τηλ.: +30 210 6785 800
Polska
Pfizer Sp. z o.o.
Tel: +48 22 335 61 00
78
-
one plaster
España
Pfizer, S.A.
Télf: +34914909900
Portugal
Laboratórios Pfizer, Lda.
Tel: (+351) 21 423 55 00
France
Pfizer
Tél:+33 1 58 07 30 00
România
Pfizer Romania S.R.L
Tel: +40 (0) 21 207 28 00
Ireland
Wyeth Pharmaceuticals
Tel:+353 1 449 3500
Slovenská republika
Pfizer Luxembourg SARL,
organiza
č
ná zložka
Tel: + 421 2 3355 5500
Ísland
Icepharma hf.
Tel:+354 540 8000
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
Italia
Wyeth Lederle S.p.A.
Tel:+39 06 927151
Sverige
Pfizer AB
Tel:+46 (0)8 550 520 00
Kύπρος
Wyeth Hellas (Cyprus Branch) AEBE
T:+357 22 817690
United Kingdom
Wyeth Pharmaceuticals
Tel:+44 845 367 0098
This leaflet was last approved in
Detailed information on this medicine is available on European Medicines Agency (EMEA) website:
79
Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/refacto_af.html
Copyright © 1995-2021 ITA all rights reserved.