










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Removab 10 microgram concentrate for solution for infusion
2.
One pre-filled syringe contains 10 microgram of catumaxomab* in 0.1 ml solution, corresponding to
0.1 mg/ml.
*rat-mouse hybrid IgG2 monoclonal antibody produced in a rat-mouse hybrid-hybridoma cell line
For a full list of excipients, see section 6.1.
3.
Concentrate for solution for infusion.
Clear and colourless solution.
4.
Removab is indicated for the intraperitoneal treatment of malignant ascites in patients with
EpCAM-positive carcinomas where standard therapy is not available or no longer feasible.
Removab must be administered under the supervision of a physician experienced in the use of
anti-neoplastic medicinal products.
Adequate monitoring of the patient after end of Removab infusion is recommended. In the pivotal
study patients were monitored for 24 h after each infusion.
Prior to the intraperitoneal infusion, pre-medication with analgesic / antipyretic / nonsteroidal
antiphlogistic medicinal products is recommended (see section 4.4).
Posology
Removab dosing schedule comprises the following four intraperitoneal infusions:
1
st
dose
10 microgram on day 0
2
nd
dose
20 microgram on day 3
3
rd
dose
50 microgram on day 7
4
th
dose
150 microgram on day 10
An interval of at least two days must elapse between infusions. The interval between the infusion days
can be prolonged in case of relevant adverse reactions. The overall treatment period should not exceed
20 days. No dose reductions of Removab were investigated during clinical trials.
Special populations
Hepatic impairment
Patients with hepatic impairment of a higher severity grade than moderate and / or with more than
70% of the liver metastasised and / or portal vein thrombosis / obstruction have not been investigated.
Treatment of these patients with Removab should only be considered after a thorough evaluation of
benefit / risk (see section 4.4).
2
Renal impairment
Patients with renal impairment of a higher severity grade than mild have not been investigated.
Treatment of these patients with Removab should only be considered after a thorough evaluation of
benefit / risk (see section 4.4).
Paediatric patients
Removab is not recommended for use in children below the age of 18 years due to a lack of data on
safety and efficacy.
Ethnicity
Patients of non-Caucasian origin have not been included in clinical studies.
Method of administration
Removab must be administered as an
intraperitoneal infusion only
.
Removab
must not
be administered by intraperitoneal bolus or by any other route of administration.
Before administration of Removab the concentrate for solution for infusion is diluted in sodium
chloride 9 mg/ml (0.9%) solution for injection. The diluted Removab solution for infusion is
administered intraperitoneally via a constant infusion pump system.
See section 6.6 for detailed instructions on dilution prior to administration and for instructions for
administration.
Hypersensitivity to the active substance or to any of the excipients.
Hypersensitivity to murine (rat and / or mouse) proteins.
Removab
must not
be administered as a bolus or by any route other than intraperitoneally.
Cytokine release related symptoms
As release of pro-inflammatory and cytotoxic cytokines is initiated by the binding of catumaxomab to
immune and tumour cells, cytokine release related clinical symptoms such as fever, nausea, vomiting
and chills have been very commonly reported during and after the Removab administration (see
section 4.8). Dyspnoea and hypo-/ hypertension are commonly observed. In the clinical studies in
patients with malignant ascites, 1000 mg paracetamol intravenously was routinely administered prior
to Removab infusion for pain and pyrexia control. Despite this premedication, patients experienced the
adverse reactions described above with an intensity of up to grade 3, according to the Common
Terminology Criteria for Adverse Events (CTCAE) of the US National Cancer Institute. Other or
additional standard pre-medication with analgesic / antipyretic / nonsteroidal antiphlogistic medicinal
products is recommended.
Systemic Inflammatory Response Syndrome (SIRS), which may also occur uncommonly due to the
mechanism of action of catumaxomab, develops, in general, within 24 hours after Removab infusion,
showing symptoms of fever, tachycardia, tachypnoea and leucocytosis (see section 4.8). Standard
therapy or premedication, e.g. analgesic / antipyretic / nonsteroidal antiphlogistic is appropriate to
limit the risk.
Abdominal pain
Abdominal pain was commonly reported as an adverse reaction. This transient effect is considered
partially a consequence of study procedures such as the intraperitoneal route of administration.
Performance status and BMI
3
A solid performance status expressed as Body Mass Index (BMI) > 17 (to be assessed after drainage of
ascites fluid) and Karnofsky Index > 60 is required prior to Removab therapy.
Acute infections
In presence of factors interfering with the immune system, in particular acute infections, the
administration of Removab is not recommended.
Ascites drainage
Appropriate medical management of ascites drainage is a prerequisite for Removab treatment in order
to assure stable circulatory and renal functions. This must at least include ascites drainage until stop of
spontaneous flow, and, if appropriate, supportive replacement therapy with crystalloids and / or
colloids. Conditions such as hypovolaemia, hypoproteinaemia, hypotension, circulatory
decompensation and acute renal impairment should be resolved prior to each Removab infusion.
Hepatic impairment or portal vein thrombosis / obstruction
Patients with hepatic impairment of a higher severity grade than moderate and / or with more than
70% of the liver metastasised and / or portal vein thrombosis / obstruction have not been investigated.
Treatment of these patients with Removab should only be considered after a thorough evaluation of
benefit / risk.
Renal impairment
Patients with renal impairment of a higher severity grade than mild have not been investigated.
Treatment of these patients with Removab should only be considered after a thorough evaluation of
benefit / risk.
Perfusion system
Only the following material must be used for the application of Removab:
•
50 ml polypropylene syringes
•
polyethylene perfusion tubing with an inner diameter of 1 mm and a length of 150 cm
•
polycarbonate infusion valves / Y connections
•
polyurethane, polyurethane silicon coated catheters
No interaction studies have been performed.
There are no adequate data from the use of Removab in pregnant women. Animal reproduction studies
have not been performed with catumaxomab. The potential risk for humans is unknown. Therefore,
Removab should not be used during pregnancy unless clearly necessary.
It is unknown whether catumaxomab is excreted in human breast milk. A decision must be made
whether to discontinue breast-feeding or to discontinue / abstain from Removab therapy taking into
account the benefit of breast-feeding for the child and the benefit of therapy for the woman.
No studies on the effects on the ability to drive and use machines have been performed.
Patients experiencing infusion-related symptoms should be advised not to drive and use machines until
symptoms abate.
The nature and frequency of adverse reactions described in this section were analysed in an integrated
safety analysis on the basis of 5 clinical studies consisting of 258 patients in the indications malignant
4
ascites (193 patients), peritoneal carcinomatosis (24 patients) and ovarian cancer (41 patients) with
intraperitoneal application of Removab.
Approximately 90% of patients experienced adverse reactions. In Table 1, adverse reactions reported
with catumaxomab are listed and classified according to frequency and System Organ Class.
Frequency groupings are defined according to the following convention: very common (≥1/10),
common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100).Within each frequency grouping,
undesirable effects are presented in order of decreasing seriousness.
Table 1 Adverse reactions with catumaxomab
Blood and lymphatic system disorders
Very common
Lymphopenia.
Common
Leucocytosis, anaemia, neutrophilia, thrombocythaemia.
Cardiac disorders
Common
Tachycardia.
Ear and labyrinth disorders
Common
Vertigo.
Gastrointestinal disorders
Very common
Abdominal pain*, nausea, vomiting, diarrhoea.
Common
Ileus*, sub-ileus*, constipation, dyspepsia, abdominal distension,
flatulence, gastric disorder, gastroesophageal reflux disease, stomatitis.
Uncommon
Gastric haemorrhage*, intestinal obstruction*.
General disorders and administration site conditions
Very common
Pyrexia*, fatigue, chills, pain.
Common
Asthenia, influenza-like illness, chest pain, oedema, thirst.
Uncommon
Application site inflammation*, extravasation*.
Hepatobiliary disorders
Common
Hyperbilirubinaemia, cytolytic hepatitis.
Infections and infestations
Common
Infection, erythaema induratum, urinary tract infection.
Uncommon
Catheter-related infection*, skin infection*.
Metabolism and nutrition disorders
Common
Anorexia, hyponatraemia, hypocalcaemia, hypokalaemia,
hypoproteinaemia, dehydration, hyperglycaemia.
Musculoskeletal and connective tissue disorders
Common
Arthralgia, back pain, myalgia.
Nervous system disorders
Common
Headache, dizziness.
Uncommon
Convulsion*.
Psychiatric disorders
Common
Anxiety, insomnia.
Renal and urinary disorders
Common
Oliguria, leucocyturia, proteinuria, haematuria.
Uncommon
Renal failure acute*.
Respiratory, thoracic and mediastinal disorders
Common
Dyspnoea*, pleural effusion.
Uncommon
Pulmonary embolism*, pleural effusion*.
Skin and subcutaneous tissue disorders
Common
Exanthema, dermatitis allergic, skin reaction, erythaema, rash,
hyperhidrosis, pruritus, urticaria.
Uncommon
Dermatitis allergic*, rash*, skin exfoliation*, skin reaction*.
Vascular disorders
Common
Hypotension, hypertension, flushing, hot flush.
* were also reported as serious adverse reactions
5










































Adverse reactions of special interest
The following definitions of CTCAE criteria of the US National Cancer Institute apply:
CTCAE grade 1 = mild, CTCAE grade 2 = moderate, CTCAE grade 3 = severe, CTCAE grade 4 =
life-threatening
Cytokine release related symptoms:
Very commonly reported acute infusion-related reactions due to release of cytokines included fever,
nausea, vomiting and chills. These reactions were frequently observed during and after Removab
infusions with a severity of grade 1 and 2 and were fully reversible. Grade 3 pyrexia (5%), vomiting
(3.9%), nausea (2.3%), dyspnoea (1.6%) hypotension (1.2%), hypertension (0.8%) and chills (0.8%)
were reported. Grade 4 dyspnoea and hypotension were also reported in one patient each. Symptoms
of pain and pyrexia can be ameliorated or avoided by pre-medication (see sections 4.2 and 4.4).
Systemic Inflammatory Response Syndrome (SIRS):
In 0.8% of the patients symptoms of SIRS were observed within 24 hours after Removab infusion,
such as grade 3 tachycardia and fever and grade 4 dyspnoea. These reactions resolved under
symptomatic treatment.
Abdominal pain:
In 48.1% of patients abdominal pain was reported as an adverse reaction reaching grade 3 in 9.7% of
patients, but it resolved under symptomatic treatment.
No case of overdose has been reported. Patients receiving a higher than recommended dose of
catumaxomab experienced more severe (grade 3) adverse reactions.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other antineoplastic agents, Monoclonal antibodies, ATC code:
L01XC09
Mechanism of action
Catumaxomab is a trifunctional rat-mouse hybrid monoclonal antibody that is specifically directed
against the epithelial cell adhesion molecule (EpCAM) and the CD3 antigen.
The EpCAM antigen is overexpressed on most carcinomas. CD3 is expressed on mature T-cells as a
component of the T-cell receptor. A third functional binding site in the Fc-region of catumaxomab
enables interaction with accessory immune cells via Fcγ receptors.
Due to
catumaxomab’s binding properties, tumour cells, T-cells and accessory immune cells come in
close proximity. Thereby, a concerted immunoreaction against tumour cells is induced which includes
different mechanisms of action such as T-cell activation, antibody-dependent cell-mediated
cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC) and phagocytosis. This results in
destruction of tumour cells.
Pharmacodynamic effects
The anti-tumour activity of catumaxomab has been demonstrated
in vitro
and
in vivo
. Effective
catumaxomab-mediated killing of tumour cells
in vitro
was observed for target cells with low and high
expression of the EpCAM antigen, independent of the primary tumour type. The
in vivo
anti-tumour
activity of catumaxomab was confirmed in an immunologically compromised mouse model of ovarian
carcinoma, where tumour development was delayed by an intraperitoneal treatment with catumaxomab
and human peripheral blood mononuclear cells.
Clinical efficacy
6
The efficacy of catumaxomab was demonstrated in a two-arm, randomised, open-label clinical trial
(IP-REM-AC-01) in 258 patients with symptomatic malignant ascites due to EpCAM-positive
carcinomas of whom 170 were randomised to catumaxomab treatment. This study compared
paracentesis plus catumaxomab
versus
paracentesis alone (control).
Catumaxomab was applied in patients where standard therapy was not available or no longer feasible
and who had a Karnofsky performance status of a least 60. Catumaxomab was administered as four
intraperitoneal infusions with increased doses of 10, 20, 50 and 150 micrograms on day 0, 3, 7 and 10,
respectively (see section 4.2). In the pivotal study IP-REM-AC-01 98.1% of patients were hospitalised
for a median of 11 days.
In this study, the primary efficacy endpoint was puncture-free survival, which was a composite
endpoint defined as the time to first need for therapeutic ascites puncture or death, whichever occurred
first. The results for puncture-free survival and time to first need for therapeutic ascites puncture in
terms of medians and hazard ratios are presented in Table 2. Kaplan Meier estimates for time to first
need for therapeutic ascites puncture are given in Figure 1.
Table 2 Efficacy results (puncture-free survival and time to first need for therapeutic ascites
puncture) of study IP-REM-AC-01 [95% CI]
Variable
Paracentesis + catumaxomab
(N=170)
Paracentesis (control)
(N=88)
Puncture free survival
Median puncture-free survival (days)
44
11
95% CI for median (days)
[31; 49]
[9; 16]
p-value
(log-rank test)
< 0.0001
Hazard ratio (HR)
0.310
95% CI for HR
[0.228; 0.423]
Time to first need for therapeutic ascites puncture
Median time to first need for therapeutic
ascites puncture (days)
77
13
95% CI for median (days)
[62;104]
[9; 17]
p-value
(log-rank test)
< 0.0001
Hazard ratio (HR)
0.169
95% CI for HR
[0.114; 0.251]
Figure 1 Kaplan-Meier estimates of time to first need for therapeutic ascites puncture of
study IP-REM-AC-01
Time (days) to event
Treatment:
Catumaxomab (N=170)
7



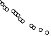












































Control (N=88)
N: number of patients in a treatment group.
The efficacy of the treatment with paracentesis and catumaxomab in patients with malignant ascites
due to EpCAM-positive carcinomas was statistically significantly superior to that with paracentesis
alone in terms of puncture-free survival and time to first need for therapeutic ascites puncture.
After completion of the study, patients were further observed until the end of their lifetime (post-study
phase) in order to assess overall survival (Table 3).
Table 3
Overall survival of study IP-REM-AC-01 in post study phase [95% CI]
Paracentesis + catumaxomab
(N=170)
Paracentesis (control)
(N=88)
Overall survival (days)
72
68
95% CI for median (days)
[61;98]
[49;81]
p-value
(log-rank test)
0.0846
Hazard ratio (HR)
0.723
95% CI for HR
[0.498; 1.048]
A positive trend for median overall survival after treatment with catumaxomab compared to control
was seen.
Immunogenicity
The induction of human anti-murine (rat and / or mouse) antibodies (HAMAs/HARAs) is an intrinsic
effect of murine monoclonal antibodies. Current data on catumaxomab derived from the pivotal study
show that only 5% of patients (7/132 patients) were HAMA positive before the 4th infusion. HAMAs
were present in 87% of patients one month after the last catumaxomab infusion. No data about clinical
effects due to the presence of HAMAs/HARAs are available to date. No hypersensitivity reactions
were observed.
5.2 Pharmacokinetic properties
Pharmacokinetics of catumaxomab during and after four intraperitoneal infusions of 10, 20, 50 and
150 microgram catumaxomab were investigated in 13 patients with symptomatic malignant ascites due
to EpCAM-positive carcinomas.
The variability between subjects was high. The geometric mean plasma C
max
was approximately 0.5
ng/ml (range 0 to 2.3), and the geometric mean plasma AUC was approximately 1.7 day* ng/ml (range
< LLOQ (lower limit of quantification) to 13.5). The geometric mean apparent terminal plasma
elimination half-life (t
1/2
) was approximately 2.5 days (range 0.7 to 17).
Catumaxomab was detectable in the ascites fluid and in plasma. The concentrations increased with the
number of infusions and the doses applied in most patients. Plasma levels tended to decline after
achieving a maximum after each dose.
Special populations
No studies have been conducted.
5.3 Preclinical safety data
Administration of catumaxomab in animal models did not result in any signs of abnormal or
drug-related acute toxicity or signs of local intolerance at the injection/infusion site. However, these
findings are of limited value due to the high species-specificity of catumaxomab.
8










Repeated-dose toxicity, genotoxicity, carcinogenicity, reproductive and developmental toxicity studies
have not been performed.
6.
6.1 List of excipients
Sodium citrate
Citric acid monohydrate
Polysorbate 80
Water for injections
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in
section 6.6.
6.3 Shelf life
24 months
After dilution
The prepared solution for infusion is physically and chemically stable for 48 hours at 2°C to 8°C and
for 24 hours at a temperature not above 25°C. From a microbiological point of view, the product
should be used immediately. If not used immediately, in-use storage times and conditions prior to use
are the responsibility of the user and would normally not be longer than 24 hours at 2°C to 8°C, unless
dilution has taken place in controlled and validated aseptic conditions.
6.4 Special precautions for storage
Store in a refrigerator (2°C-8°C). Do not freeze. Store in the original package in order to protect from
light.
For storage conditions of the diluted medicinal product, see section 6.3.
6.5 Nature and contents of container
0.1 ml concentrate for solution for infusion in a pre-filled syringe (type I glass, siliconised) with
plunger stopper (bromobutyl rubber) and luer lock system (polypropylene siliconised and
polycarbonate) with tip cap (styrene butadiene rubber) with a cannula; pack size of 1.
6.6 Special precautions for disposal and other handling
Disposal
No special requirements.
Material and equipment required
The following components must be used for the dilution and administration of Removab as Removab
is only compatible with:
•
50 ml polypropylene syringes
•
polyethylene perfusion tubings with an inner diameter of 1 mm and a length of 150 cm
•
polycarbonate infusion valves / Y connections
•
polyurethane, polyurethane silicon coated catheters
In addition the following is required:
9
•
Sodium chloride 9 mg/ml (0.9%) solution for injection
•
Precision perfusion pump
Instructions for dilution prior to administration
Removab should be prepared by a healthcare professional using appropriate aseptic technique.
The outer surface of the pre-filled syringe is not sterile.
•
Based on the dose, the appropriate amount of sodium chloride 9 mg/ml (0.9%) solution for
injection is extracted with a 50 ml syringe (Table 4).
•
An additional air buffer of at least 3 ml is included in the 50 ml syringe.
•
The tip cap from the Removab pre-filled syringe is removed with the tip pointing up.
•
The enclosed cannula is attached to the Removab pre-filled syringe. For each syringe a new
cannula is used.
•
The pre-filled syringe cannula is inserted through the 50 ml syringe opening so that the cannula
is immersed in the sodium chloride 9 mg/ml (0.9%) solution for injection (Figure 2).
•
The entire content of the syringe (Removab concentrate plus air buffer) is injected from the
pre-filled syringe directly into the sodium chloride 9 mg/ml (0.9%) solution for injection.
•
The plunger rod MUST NOT be drawn back to rinse the pre-filled syringe, in order to avoid
contamination and to ensure that the correct volume is ejected.
•
The 50 ml syringe is closed with a cap and shaken gently to mix the solution. Any air bubble(s)
from the 50 ml syringe is eliminated.
•
The peelable sticker, which is provided on the inner side of the Removab carton box, displaying
the text “Diluted Removab. Intraperitoneal use only.” must be attached to the 50 ml syringe
containing the diluted Removab solution for intraperitoneal infusion. This is a precautionary
measure to ensure that Removab is infused only via the intraperitoneal route of administration.
•
The 50 ml syringe is inserted in the infusion pump.
Table 4 Preparation of Removab solution for intraperitoneal infusion
Number of
infusion /
Dose
Number of Removab pre-filled
syringe(s)
Total volume
of Removab
concentrate for
solution for
infusion
Sodium
chloride
9 mg/ml
(0.9%) solution
for injection
Final volume
for
administration
10 microgram
pre-filled
syringe
50 microgram
pre-filled
syringe
1
st
infusion
10 microgram
1
0.1 ml
10 ml
10.1 ml
2
nd
infusion
20 microgram
2
0.2 ml
20 ml
20.2 ml
3
rd
infusion
50 microgram
1
0.5 ml
49.5 ml
50 ml
4
th
infusion
150 microgram
3
1.5 ml
48.5 ml
50 ml
10
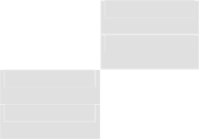















Figure 2 Illustration of the transfer of Removab from the pre-filled syringe to the 50 ml
syringe
Pre-filled syringe
Air buffer
Removab solution
Air buffer
Sodium Chloride 9 mg/ml (0.9%)
50 ml Syringe
Method of administration:
The catheter for intraperitoneal administration should be placed under ultrasound guidance by a
physician experienced in intraperitoneal administration procedures. The catheter is used for ascites
drainage and infusion of diluted Removab and sodium chloride 9 mg/ml (0.9%) solution for injection.
It is recommended that the catheter remains in the abdominal cavity during the entire treatment period.
It can be removed the day after the last infusion.
Prior to each Removab administration the ascites fluid must be drained until stop of spontaneous flow
(see section 4.4). Subsequently, prior to each Removab administration 500 ml sodium chloride
9 mg/ml (0.9%) solution for injection shall be infused to support distribution of the antibody in the
abdominal cavity.
Removab must be administered intraperitoneally over 6 hours via a constant infusion pump system as
described below:
•
The 50 ml syringe containing the diluted Removab solution for infusion is installed in the
precision pump.
•
The connected perfusion tubing equipment of the precision pump is prefilled with the diluted
Removab solution for infusion. A perfusion tubing of an inner diameter of 1 mm and a length of
150 cm must be used.
•
The perfusion tubing is connected to the Y-connection.
•
Parallel to each Removab application 250 ml sodium chloride 9 mg/ml (0.9%) solution for
injection are infused via an infusion valve / Y connection in the perfusion lead of the catheter.
•
The pump speed is adjusted according to the volume to be administered and the infusion time of
6 hours.
•
After completion of the Removab infusion 20 ml sodium chloride 9 mg/ml (0.9%) solution for
injection are infused briefly to clear the dead volume in the perfusion lead.
•
The catheter is kept closed until the next infusion.
•
The day after the last infusion a drainage of ascites until stop of spontaneous flow
is performed.
Subsequently, the catheter can be removed.
11



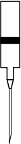




Figure 3 Schematic illustration of the infusion system
250 ml
Sodium Chloride 9 mg/ml (0.9%)
Infusion
valve
Perfusion
Lead
Catheter
Removab solution
for i.p. infusion
Perfusion Tubing
(1 mm inner diameter, 150 cm length)
7.
Fresenius Biotech GmbH
Am Haag 6-7
82166 Graefelfing
Germany
Tel: +49 (0)6172 608-2240
8.
EU/1/09/512/001
9.
20/04/2009
Detailed information on this medicine is available on the European Medicines Agency (EMEA) web site:
http://www.emea.europa.eu/
.
12




1.
Removab 50 microgram concentrate for solution for infusion
2.
One pre-filled syringe contains 50 microgram of catumaxomab* in 0.5 ml solution, corresponding to
0.1 mg/ml.
*rat-mouse hybrid IgG2 monoclonal antibody produced in a rat-mouse hybrid-hybridoma cell line
For a full list of excipients, see section 6.1.
3.
Concentrate for solution for infusion.
Clear and colourless solution.
4.
Removab is indicated for the intraperitoneal treatment of malignant ascites in patients with
EpCAM-positive carcinomas where standard therapy is not available or no longer feasible.
Removab must be administered under the supervision of a physician experienced in the use of
anti-neoplastic medicinal products.
Adequate monitoring of the patient after end of Removab infusion is recommended. In the pivotal
study patients were monitored for 24 h after each infusion.
Prior to the intraperitoneal infusion pre-medication with analgesic / antipyretic / nonsteroidal
antiphlogistic medicinal products is recommended (see section 4.4).
Posology
Removab dosing schedule comprises the following four intraperitoneal infusions:
1
st
dose
10 microgram on day 0
2
nd
dose
20 microgram on day 3
3
rd
dose
50 microgram on day 7
4
th
dose
150 microgram on day 10
An interval of at least two days must elapse between infusions. The interval between the infusion days
can be prolonged in case of relevant adverse reactions. The overall treatment period should not exceed
20 days. No dose reductions of Removab were investigated during clinical trials.
Special populations
Hepatic impairment
Patients with hepatic impairment of a higher severity grade than moderate and / or with more than
70% of the liver metastasised and / or portal vein thrombosis / obstruction have not been investigated.
Treatment of these patients with Removab should only be considered after a thorough evaluation of
benefit / risk (see section 4.4).
13
Renal impairment
Patients with renal impairment of a higher severity grade than mild have not been investigated.
Treatment of these patients with Removab should only be considered after a thorough evaluation of
benefit / risk (see section 4.4).
Paediatric patients
Removab is not recommended for use in children below the age of 18 years due to a lack of data on
safety and efficacy.
Ethnicity
Patients of non-Caucasian origin have not been included in clinical studies.
Method of administration
Removab must be administered as an
intraperitoneal infusion only
.
Removab
must not
be administered by intraperitoneal bolus or by any other route of administration.
Before administration of Removab the concentrate for solution for infusion is diluted in sodium
chloride 9 mg/ml (0.9%) solution for injection. The diluted Removab solution for infusion is
administered intraperitoneally via a constant infusion pump system.
See section 6.6 for detailed instructions on dilution prior to administration and for instructions for
administration.
Hypersensitivity to the active substance or to any of the excipients.
Hypersensitivity to murine (rat and / or mouse) proteins.
Removab
must not
be administered as a bolus or by any route other than intraperitoneally.
Cytokine release related symptoms
As release of pro-inflammatory and cytotoxic cytokines is initiated by the binding of catumaxomab to
immune and tumour cells, cytokine release related clinical symptoms such as fever, nausea, vomiting
and chills have been very commonly reported during and after the Removab administration (see
section 4.8). Dyspnoea and hypo-/ hypertension are commonly observed. In the clinical studies in
patients with malignant ascites, 1000 mg paracetamol intravenously was routinely administered prior
to Removab infusion for pain and pyrexia control. Despite this premedication, patients experienced the
adverse reactions described above with an intensity of up to grade 3, according to the Common
Terminology Criteria for Adverse Events (CTCAE) of the US National Cancer Institute. Other or
additional standard pre medication with analgesic / antipyretic / nonsteroidal antiphlogistic medicinal
products is recommended.
Systemic Inflammatory Response Syndrome (SIRS), which may also occur uncommonly due to the
mechanism of action of catumaxomab, develops, in general, within 24 hours after Removab infusion,
showing symptoms of fever, tachycardia, tachypnoea and leucocytosis (see section 4.8). Standard
therapy or premedication, e.g. analgesic / antipyretic / non-steroidal antiphlogistic is appropriate to
limit the risk.
Abdominal pain
Abdominal pain was commonly reported as an adverse reaction. This transient effect is considered
partially a consequence of study procedures such as the intraperitoneal route of administration.
Performance status and BMI
14
A solid performance status expressed as Body Mass Index (BMI) >17 (to be assessed after drainage of
ascites fluid) and Karnofsky Index > 60 is required prior to Removab therapy.
Acute infections
In presence of factors interfering with the immune system, in particular acute infections, the
administration of Removab is not recommended.
Ascites drainage
Appropriate medical management of ascites drainage is a prerequisite for Removab treatment in order
to assure stable circulatory and renal functions. This must at least include ascites drainage until stop of
spontaneous flow, and if appropriate supportive replacement therapy with crystalloids and / or
colloids. Conditions such as hypovolaemia, hypoproteinaemia, hypotension, circulatory
decompensation and acute renal impairment should be resolved prior to each Removab infusion.
Hepatic impairment or portal vein thrombosis / obstruction
Patients with hepatic impairment of a higher severity grade than moderate and / or with more than
70% of the liver metastasised and / or portal vein thrombosis / obstruction have not been investigated.
Treatment of these patients with Removab should only be considered after a thorough evaluation of
benefit / risk.
Renal impairment
Patients with renal impairment of a higher severity grade than mild have not been investigated.
Treatment of these patients with Removab should only be considered after a thorough evaluation of
benefit / risk.
Perfusion system
Only the following material must be used for the application of Removab:
•
50 ml polypropylene syringes
•
polyethylene perfusion tubing with an inner diameter of 1 mm and a length of 150 cm
•
polycarbonate infusion valves / Y connections
•
polyurethane, polyurethane silicon coated catheters
No interaction studies have been performed.
There are no adequate data from the use of Removab in pregnant women. Animal reproduction studies
have not been performed with catumaxomab. The potential risk for humans is unknown. Therefore,
Removab should not be used during pregnancy unless clearly necessary.
It is unknown whether catumaxomab is excreted in human breast milk. A decision must be made
whether to discontinue breast-feeding or to discontinue / abstain from Removab therapy, taking into
account the benefit of breast-feeding for the child and the benefit of therapy for the woman.
No studies on the effects on the ability to drive and use machines have been performed.
Patients experiencing infusion-related symptoms should be advised not to drive and use machines until
symptoms abate.
The nature and frequency of adverse reactions described in this section were analysed in an integrated
safety analysis on the basis of 5 clinical studies consisting of 258 patients in the indications malignant
15
ascites (193 patients), peritoneal carcinomatosis (24 patients) and ovarian cancer (41 patients) with
intraperitoneal application of Removab.
Approximately 90% of patients experienced adverse reactions. In Table 1, adverse reactions reported
with catumaxomab are listed and classified according to frequency and System Organ Class.
Frequency groupings are defined according to the following convention: very common (≥1/10),
common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Table 1 Adverse reactions with catumaxomab
Blood and lymphatic system disorders
Very common
Lymphopenia.
Common
Leucocytosis, anaemia, neutrophilia, thrombocythaemia.
Cardiac disorders
Common
Tachycardia.
Ear and labyrinth disorders
Common
Vertigo.
Gastrointestinal disorders
Very common
Abdominal pain*, nausea, vomiting, diarrhoea.
Common
Ileus*, sub-ileus*, constipation, dyspepsia, abdominal distension,
flatulence, gastric disorder, gastroesophageal reflux disease, stomatitis.
Uncommon
Gastric haemorrhage*, intestinal obstruction*.
General disorders and administration site conditions
Very common
Pyrexia*, fatigue, chills, pain.
Common
Asthenia, influenza-like illness, chest pain, oedema, thirst.
Uncommon
Application site inflammation*, extravasation*.
Hepatobiliary disorders
Common
Hyperbilirubinaemia, cytolytic hepatitis.
Infections and infestations
Common
Infection, erythaema induratum, urinary tract infection.
Uncommon
Catheter-related infection*, skin infection*.
Metabolism and nutrition disorders
Common
Anorexia, hyponatraemia, hypocalcaemia, hypokalaemia,
hypoproteinaemia, dehydration, hyperglycaemia.
Musculoskeletal and connective tissue disorders
Common
Arthralgia, back pain, myalgia.
Nervous system disorders
Common
Headache, dizziness.
Uncommon
Convulsion*.
Psychiatric disorders
Common
Anxiety, insomnia.
Renal and urinary disorders
Common
Oliguria, leucocyturia, proteinuria, haematuria.
Uncommon
Renal failure acute*.
Respiratory, thoracic and mediastinal disorders
Common
Dyspnoea*, pleural effusion.
Uncommon
Pulmonary embolism*, pleural effusion*.
Skin and subcutaneous tissue disorders
Common
Exanthema, dermatitis allergic, skin reaction, erythaema, rash,
hyperhidrosis, pruritus, urticaria.
Uncommon
Dermatitis allergic*, rash*, skin exfoliation*, skin reaction*.
Vascular disorders
Common
Hypotension, hypertension, flushing, hot flush.
* were also reported as serious adverse reactions
16










































Adverse reactions of special interest
The following definitions of CTCAE criteria of the US National Cancer Institute apply:
CTCAE grade 1 = mild, CTCAE grade 2 = moderate, CTCAE grade 3 = severe, CTCAE grade 4 =
life-threatening
Cytokine release related symptoms:
Very commonly reported acute infusion-related reactions due to release of cytokines included fever,
nausea, vomiting and chills. These reactions were frequently observed during and after Removab
infusions with a severity of grade 1 and 2 and were fully reversible. Grade 3 pyrexia (5%), vomiting
(3.9%), nausea (2.3%), dyspnoea (1.6%), hypotension (1.2%), hypertension (0.8%) and chills (0.8%)
were reported. Grade 4 dyspnoea and hypotension were also reported in one patient each. Symptoms
of pain and pyrexia can be ameliorated or avoided by pre-medication (see sections 4.2 and 4.4).
Systemic Inflammatory Response Syndrome (SIRS):
In 0.8% of the patients symptoms of SIRS were observed within 24 hours after Removab infusion,
such as grade 3 tachycardia and fever and grade 4 dyspnoea. These reactions resolved under
symptomatic treatment.
Abdominal pain:
In 48.1% of patients abdominal pain was reported as an adverse reaction reaching grade 3 in 9.7% of
patients, but it resolved under symptomatic treatment.
No case of overdose has been reported. Patients receiving a higher than recommended dose of
catumaxomab experienced more severe (grade 3) adverse reactions.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other antineoplastic agents, Monoclonal antibodies, ATC code:
L01XC09
Mechanism of action
Catumaxomab is a rat-mouse hybrid monoclonal trifunctional antibody that is specifically directed
against the epithelial cell adhesion molecule (EpCAM) and the CD3 antigen.
The EpCAM antigen is overexpressed on most carcinomas. CD3 is expressed on mature T-cells as a
component of the T-cell receptor. A third functional binding site in the Fc-region of catumaxomab
enables interaction with accessory immune cells via Fcγ receptors.
Due to
catumaxomab’s binding properties, tumour cells, T-cells and accessory immune cells come in
close proximity. Thereby, a concerted immunoreaction against tumour cells is induced which includes
different mechanisms of action such as T-cell activation, antibody-dependent cell-mediated
cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC) and phagocytosis. This results in
destruction of tumour cells.
Pharmacodynamic effects
The anti-tumour activity of catumaxomab has been demonstrated
in vitro
and
in vivo
. Effective
catumaxomab-mediated killing of tumour cells
in vitro
was observed for target cells with low and high
expression of the EpCAM antigen, independent of the primary tumour type. The
in vivo
anti-tumour
activity of catumaxomab was confirmed in an immunologically compromised mouse model of ovarian
carcinoma, where tumour development was delayed by an intraperitoneal treatment with catumaxomab
and human peripheral blood mononuclear cells.
Clinical efficacy
17
The efficacy of catumaxomab was demonstrated in a two-arm, randomised, open-label clinical trial
(IP-REM-AC-01) in 258 patients with symptomatic malignant ascites due to EpCAM-positive
carcinomas of whom 170 were randomised to catumaxomab treatment. This study compared
paracentesis plus catumaxomab
versus
paracentesis alone (control).
Catumaxomab was applied in patients where standard therapy was not available or no longer feasible
and who had a Karnofsky performance status of a least 60. Catumaxomab was administered as four
intraperitoneal infusions with increased doses of 10, 20, 50 and 150 micrograms on day 0, 3, 7 and 10,
respectively (see section 4.2). In the pivotal study IP-REM-AC-01 98.1% of patients were hospitalised
for a median of 11 days.
In this study, the primary efficacy endpoint was puncture-free survival, which was a composite
endpoint defined as the time to first need for therapeutic ascites puncture or death, whichever occurred
first. The results for puncture-free survival and time to first need for therapeutic ascites puncture in
terms of medians and hazard ratios are presented in Table 2. Kaplan Meier estimates for time to first
need for therapeutic ascites puncture are given in Figure 1.
Table 2 Efficacy results (puncture-free survival and time to first need for therapeutic ascites
puncture) of study IP-REM-AC-01 [95% CI]
Variable
Paracentesis + catumaxomab
(N=170)
Paracentesis (control)
(N=88)
Puncture free survival
Median puncture-free survival (days)
44
11
95% CI for median (days)
[31; 49]
[9; 16]
p-value
(log-rank test)
< 0.0001
Hazard ratio (HR)
0.310
95% CI for HR
[0.228; 0.423]
Time to first need for therapeutic ascites puncture
Median time to first need for therapeutic
ascites puncture (days)
77
13
95% CI for median (days)
[62;104]
[9; 17]
p-value
(log-rank test)
< 0.0001
Hazard ratio (HR)
0.169
95% CI for HR
[0.114; 0.251]
Figure 1 Kaplan-Meier estimates of time to first need for therapeutic ascites puncture of
study IP-REM-AC-01
Time (days) to event
Treatment:
Catumaxomab (N=170)
18



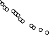












































Control (N=88)
N: number of patients in a treatment group.
The efficacy of the treatment with paracentesis and catumaxomab in patients with malignant ascites
due to EpCAM-positive carcinomas was statistically significantly superior to that with paracentesis
alone in terms of puncture-free survival and time to first need for therapeutic ascites puncture.
After completion of the study, patients were further observed until the end of their lifetime (post-study
phase) in order to assess overall survival (Table 3).
Table 3
Overall survival of study IP-REM-AC-01 in post study phase [95% CI]
Paracentesis + catumaxomab
(N=170)
Paracentesis (control)
(N=88)
Overall survival (days)
72
68
95% CI for median (days)
[61;98]
[49;81]
p-value
(log-rank test)
0.0846
Hazard ratio (HR)
0.723
95% CI for HR
[0.498; 1.048]
A positive trend for median overall survival after treatment with catumaxomab compared to control
was seen.
Immunogenicity
The induction of human anti-murine (rat and / or mouse) antibodies (HAMAs/HARAs) is an intrinsic
effect of murine monoclonal antibodies. Current data on catumaxomab derived from the pivotal study
show that only 5% of patients (7/132 patients) were HAMA positive before the 4th infusion. HAMAs
were present in 87% of patients one month after the last catumaxomab infusion. No data about clinical
effects due to the presence of HAMAs/HARAs are available to date. No hypersensitivity reactions
were observed.
5.2 Pharmacokinetic properties
Pharmacokinetics of catumaxomab during and after four intraperitoneal infusions of 10, 20, 50 and
150 microgram catumaxomab were investigated in 13 patients with symptomatic malignant ascites due
to EpCAM-positive carcinomas.
The variability between subjects was high. The geometric mean plasma C
max
was approximately
0.5 ng/ml (range 0 to 2.3) and the geometric mean plasma AUC was approximately 1.7 day*ng/ml
(range < LLOQ (lower limit of quantification) to 13.5). The geometric mean apparent terminal plasma
elimination half-life (t
1/2
) was approximately 2.5 days (range 0.7 to 17).
Catumaxomab was detectable in the ascites fluid and in plasma. The concentrations increased with the
number of infusions and the doses applied in most patients. Plasma levels tended to decline after
achieving a maximum after each dose.
Special populations
No studies have been conducted.
5.3 Preclinical safety data
Administration of catumaxomab in animal models did not result in any signs of abnormal or
drug-related acute toxicity or signs of local intolerance at the injection/infusion site. However, these
findings are of limited value due to the high species-specificity of catumaxomab.
19










Repeated-dose toxicity, genotoxicity, carcinogenicity, reproductive and developmental toxicity studies
have not been performed.
6.
6.1 List of excipients
Sodium citrate
Citric acid monohydrate
Polysorbate 80
Water for injections
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in
section 6.6.
6.3 Shelf life
24 months
After dilution
The prepared solution for infusion is physically and chemically stable for 48 hours at 2°C to 8°C and
for 24 hours at a temperature not above 25°C. From a microbiological point of view, the product
should be used immediately. If not used immediately, in-use storage times and conditions prior to use
are the responsibility of the user and would normally
not be longer than 24 hours at 2°C to 8°C, unless
dilution has taken place in controlled and validated aseptic conditions.
6.4 Special precautions for storage
Store in a refrigerator (2°C-8°C). Do not freeze. Store in the original package in order to protect from
light.
For storage conditions of the diluted medicinal product, see section 6.3.
6.5 Nature and contents of container
0.5 ml concentrate for solution for infusion in a pre-filled syringe (type I glass, siliconised) with
plunger stopper (bromobutyl rubber) and luer lock system (polypropylene siliconised and
polycarbonate) with tip cap (styrene butadiene rubber) with a cannula; pack size of 1.
6.6 Special precautions for disposal and other handling
Disposal
No special requirements.
Material and equipment required
The following components must be used for the dilution and administration of Removab as Removab
is only compatible with:
•
50 ml polypropylene syringes
•
polyethylene perfusion tubings with an inner diameter of 1 mm and a length of 150 cm
•
polycarbonate infusion valves / Y connections
•
polyurethane, polyurethane silicon coated catheters
In addition the following is required:
20
•
Sodium chloride 9 mg/ml (0.9%) solution for injection
•
Precision perfusion pump
Instructions for dilution prior to administration
Removab should be prepared by a healthcare professional using appropriate aseptic technique.
The outer surface of the pre-filled syringe is not sterile.
•
Based on the dose, the appropriate amount of sodium chloride 9 mg/ml (0.9%) solution for
injection is extracted with a 50 ml syringe (Table 4).
•
An additional air buffer of at least 3 ml is included in the 50 ml syringe.
•
The tip cap from the Removab pre-filled syringe is removed with the tip pointing up.
•
The enclosed cannula is attached to the Removab pre-filled syringe. For each syringe a new
cannula is used.
•
The pre-filled syringe cannula is inserted through the 50 ml syringe opening so that the cannula
is immersed in the sodium chloride 9 mg/ml (0.9%) solution for injection (Figure 2).
•
The entire content of the syringe (Removab concentrate plus air buffer) is injected from the
pre-filled syringe directly into the sodium chloride 9 mg/ml (0.9%) solution for injection.
•
The plunger rod MUST NOT be drawn back to rinse the pre-filled syringe, in order to avoid
contamination and to ensure that the correct volume is ejected.
•
The 50 ml syringe is closed with a cap and shaken gently to mix the solution. Any air bubble(s)
from the 50 ml syringe is eliminated.
•
The peelable sticker, which is provided on the inner side of the Removab carton box, displaying
the text “Diluted Removab. Intraperitoneal use only.” must be attached to the 50 ml syringe
containing the diluted Removab solution for intraperitoneal infusion. This is a precautionary
measure to ensure that Removab is infused only via the intraperitoneal route of administration.
•
The 50 ml syringe is inserted in the infusion pump.
Table 4 Preparation of Removab solution for intraperitoneal infusion
Number of
infusion /
Dose
Number of Removab pre-filled
syringe(s)
Total volume
of Removab
concentrate for
solution for
infusion
Sodium
chloride
9 mg/ml
(0.9%) solution
for injection
Final volume
for
administration
10 microgram
pre-filled
syringe
50 microgram
pre-filled
syringe
1
st
infusion
10 microgram
1
0.1 ml
10 ml
10.1 ml
2
nd
infusion
20 microgram
2
0.2 ml
20 ml
20.2 ml
3
rd
infusion
50 microgram
1
0.5 ml
49.5 ml
50 ml
4
th
infusion
150 microgram
3
1.5 ml
48.5 ml
50 ml
21















Figure 2 Illustration of the transfer of Removab from the pre-filled syringe to the 50 ml
syringe
Pre-filled syringe
Air buffer
Removab solution
Air buffer
Sodium Chloride 9 mg/ml (0.9%)
50 ml Syringe
Method of administration:
The catheter for intraperitoneal administration should be placed under ultrasound guidance by a
physician experienced in intraperitoneal administration procedures. The catheter is used for ascites
drainage and infusion of diluted Removab and sodium chloride 9 mg/ml (0.9%) solution for injection.
It is recommended that the catheter remains in the abdominal cavity during the entire treatment period.
It can be removed the day after the last infusion.
Prior to each Removab administration the ascites fluid must be drained until stop of spontaneous flow
(see section 4.4). Subsequently, prior to each Removab administration 500 ml sodium chloride
9 mg/ml (0.9%) solution for injection shall be infused to support distribution of the antibody in the
abdominal cavity.
Removab must be administered intraperitoneally over 6 hours via a constant infusion pump system as
described below:
•
The 50 ml syringe containing the diluted Removab solution for infusion is installed in the
precision pump.
•
The connected perfusion tubing equipment of the precision pump is prefilled with the diluted
Removab solution for infusion. A perfusion tubing of an inner diameter of 1 mm and a length of
150 cm must be used.
•
The perfusion tubing is connected to the Y-connection.
•
Parallel to each Removab application 250 ml sodium chloride 9 mg/ml (0.9%) solution for
injection are infused via an infusion valve / Y connection in the perfusion lead of the catheter.
•
The pump speed is adjusted according to the volume to be administered and the infusion time of
6 hours.
•
After completion of the Removab infusion 20 ml sodium chloride 9 mg/ml (0.9%) solution for
injection are infused briefly to clear the dead volume in the perfusion lead.
•
The catheter is kept closed until the next infusion.
•
The day after the last infusion a drainage of ascites until stop of spontaneous flow
is performed.
Subsequently, the catheter can be removed.
22







Figure 3 Schematic illustration of the infusion system
250 ml
Sodium Chloride 9 mg/ml (0.9%)
Infusion
valve
Perfusion
Lead
Catheter
Removab solution
for i.p. infusion
Perfusion Tubing
(1 mm inner diameter, 150 cm length)
7.
Fresenius Biotech GmbH
Am Haag 6-7
82166 Graefelfing
Germany
Tel: +49 (0)6172 608-2240
8.
EU/1/09/512/002
9.
20/04/2009
Detailed information on this medicine is available on the European Medicines Agency (EMEA) web site:
http://www.emea.europa.eu/
.
23




ANNEX II
A.
MANUFACTURERS OF THE BIOLOGICAL ACTIVE
SUBSTANCE AND MANUFACTURING AUTHORISATION
HOLDER RESPONSIBLE FOR BATCH RELEASE
B.
CONDITIONS OF THE MARKETING AUTHORISATION
24
A. MANUFACTURERS OF THE BIOLOGICAL ACTIVE SUBSTANCE AND
MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturers of the biological active substance
Trion Pharma GmbH
Frankfurter Ring 193a
DE-80807 Munich
Germany
Name and address of the manufacturer responsible for batch release
Fresenius Biotech GmbH
Am Haag 6-7
82166 Graefelfing
Germany
B. CONDITIONS OF THE MARKETING AUTHORISATION
Medicinal product subject to restricted medical prescription (See Annex I: Summary of Product
Characteristics, section 4.2).
•
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND
EFFECTIVE USE OF THE MEDICINAL PRODUCT
Not applicable.
•
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, as described in version 3.1 presented in
Module 1.8.1. of the Marketing Authorisation Application, is in place and functioning before and
whilst the product is on the market.
Risk Management Plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in
the Pharmacovigilance Plan, as agreed in version 5.0 of the Risk Management Plan (RMP) presented
in Module 1.8.2. of the Marketing Authorisation Application and any subsequent updates of the RMP
agreed by the CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, the
updated RMP should be submitted at the same time as the next Periodic Safety Update Report
(PSUR).
In addition, an updated RMP should be submitted
•
When new information is received that may impact on the current Safety Specification,
Pharmacovigilance Plan or risk minimisation activities
•
Within 60 days of an important (pharmacovigilance or risk minimisation) milestone being
reached
•
At the request of the EMEA.
25
ANNEX III
LABELLING AND PACKAGE LEAFLET
26
A. LABELLING
27
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
Carton: Removab 10 microgram
1.
Removab 10 microgram concentrate for solution for infusion
catumaxomab
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
One pre-filled syringe contains 10 microgram catumaxomab in 0.1 ml solution, corresponding to
0.1 mg/ml.
3.
LIST OF EXCIPIENTS
Sodium citrate, citric acid monohydrate, polysorbate 80, water for injections
4.
Concentrate for solution for infusion.
1 pre-filled syringe.
1 sterile cannula
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Intraperitoneal use only, after dilution.
Read the package leaflet before use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator. Do not freeze. Store in the original package in order to protect from light.
28























10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Fresenius Biotech GmbH
Am Haag 6-7
82166 Graefelfing
Germany
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Justification for not including Braille accepted
29


















MINIMUM PARTICULARS TO APPEAR ON BLISTERS OR STRIPS
Blister: Removab 10 microgram
1.
Removab 10 microgram concentrate for solution for infusion
catumaxomab
2.
Fresenius Biotech GmbH
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
1 pre-filled syringe.
Intraperitoneal use only, after dilution. Read the package leaflet before use.
Store in a refrigerator. Do not freeze. Store in the original package in order to protect from light.
30













MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
Pre-filled syringe: Removab 10 microgram
1.
Removab 10 microgram concentrate for solution for infusion
catumaxomab
Intraperitoneal use only, after dilution.
2.
METHOD OF ADMINISTRATION
Read the package leaflet before use.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
0.1 ml
6.
OTHER
Fresenius Biotech GmbH
31

















PARTICULARS TO APPEAR ON THE OUTER PACKAGING
Carton: Removab 50 microgram
1.
Removab 50 microgram concentrate for solution for infusion
catumaxomab
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
One pre-filled syringe contains 50 microgram catumaxomab in 0.5 ml solution, corresponding to
0.1 mg/ml.
3.
LIST OF EXCIPIENTS
Sodium citrate, citric acid monohydrate, polysorbate 80, water for injections
4.
Concentrate for solution for infusion.
1 pre-filled syringe.
1 sterile cannula
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Intraperitoneal use only, after dilution.
Read the package leaflet before use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator. Do not freeze. Store in the original package in order to protect from light.
32























10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Fresenius Biotech GmbH
Am Haag 6-7
82166 Graefelfing
Germany
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Justification for not including Braille accepted
33


















MINIMUM PARTICULARS TO APPEAR ON BLISTERS OR STRIPS
Blister: Removab 50 microgram
1.
Removab 50 microgram concentrate for solution for infusion
catumaxomab
2.
Fresenius Biotech GmbH
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
1 pre-filled syringe.
Intraperitoneal use only, after dilution. Read the package leaflet before use.
Store in a refrigerator. Do not freeze. Store in the original package in order to protect from light.
34













MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
Pre-filled syringe: Removab 50 microgram
1.
Removab 50 microgram concentrate for solution for infusion
catumaxomab
Intraperitoneal use only, after dilution.
2.
METHOD OF ADMINISTRATION
Read the package leaflet before use.
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
0.5 ml
6.
OTHER
Fresenius Biotech GmbH
35

















WARNING TEXT FOR PEELABLE STICKER TO BE ATTACHED TO 50ml SYRINGE
CONTAINING THE DILUTED REMOVAB SOLUTION FOR INFUSION
(Part of the Outer Carton)
Diluted Removab.
Intraperitoneal use only.
36



B. PACKAGE LEAFLET
37
PACKAGE LEAFLET: INFORMATION FOR THE USER
Removab 10 microgram concentrate for solution for infusion
catumaxomab
Read all of this leaflet carefully before you start using this medicine.
-
Keep this leaflet. You may need to read it again.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
-
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor.
In this leaflet:
1.
What Removab is and what it is used for
2.
Before you use Removab
3.
How to use Removab
4.
How to store Removab
6.
Further information
1.
WHAT REMOVAB IS AND WHAT IT IS USED FOR
Removab contains the active substance catumaxomab, a monoclonal antibody. It recognises a protein
on the surface of cancer cells and recruits immune cells to destroy them.
Removab is used to treat malignant ascites,
when standard treatment is not available or no longer
feasible. Malignant ascites is an accumulation of fluid in the abdominal space (peritoneal cavity)
resulting from certain types of cancer.
2.
BEFORE YOU USE REMOVAB
Do not use Removab
-
if you are allergic (hypersensitive) to catumaxomab or any of the other ingredients of Removab
(see section 6)
-
if you are allergic (hypersensitive) to murine proteins (from rat and / or mouse)
Take special care with Removab
It is important to tell your doctor
if you have any of the following:
-
undrained fluid in your abdominal cavity
-
cold hands and feet, light headedness, difficulty passing urine, increased heart rate, and
weakness (symptoms of low blood volume)
-
weight gain, weakness, shortness of breath and fluid retention (symptoms of low blood protein
levels)
-
feeling dizzy and faint (symptoms of low blood pressure)
-
problems with your heart and circulation
-
kidney or liver problems
Removab should not be used in children and adolescents under 18 years of age.
Before you start using Removab your doctor will check your:
-
Body Mass Index (BMI), which depends on your height and weight
38
-
If you have any further questions, ask your doctor.
5.
Possible side effects
-
an infection.
- Karnofsky Index, a measure of your general performance status.
You are required to have a BMI above 17 (after drainage of the ascites fluid) and a Karnofsky Index
above 60 to use this medicine.
Taking other medicines
Please tell your doctor or pharmacist if you are taking or have recently taken any other medicines
including those obtained without a prescription.
Pregnancy and breast-feeding
You should not use Removab if you are pregnant unless clearly necessary. Talk to your doctor if you
are, might be or are planning to become pregnant.
If you are breast-feeding, talk to your doctor before starting treatment.
Driving and using machines
There are no studies on the effects of Removab on the ability to drive and use machines.However, if
you experience side effects such as dizziness or chills during or after administration, you should not
drive or use machines until they disappear.
3.
HOW TO USE REMOVAB
You will be given Removab under the supervision of a doctor experienced in treating cancer. After the
Removab infusion you will be observed as decided by your doctor.
Before starting and during treatment, you will be given other medicines to reduce fever, pain or
inflammation caused by Removab.
A catheter will be placed in your abdominal space (intraperitoneal) for the whole treatment period,
until the day after your last infusion.
Removab is given as 4 intraperitoneal infusions with increasing dose (10, 20, 50 and 150 micrograms),
separated at least by a 2-day break.
If you have any further questions on the use of this product, ask your doctor.
4.
POSSIBLE SIDE EFFECTS
Like all medicines, Removab can cause side effects, although not everybody gets them.
These side effects may occur with certain frequencies, which are defined as follows:
very common: affects more than 1 user in 10
common: affects 1 to 10 users in 100
uncommon: affects 1 to 10 users in 1,000
rare: affects 1 to 10 users in 10,000
very rare: affects less than 1 user in 10,000
not known: frequency cannot be estimated from the available data.
Serious side effects
Some of these side effects may be serious and require medical treatment. You should tell a doctor
immediately if you experience any of these serious side effects.
Very common serious side effects:
-
Abdominal pain
-
Fever
39
Common serious side effects:
-
Abdominal pain accompanied by difficulty passing stools
-
Shortness of breath
Uncommon serious side effects:
-
Very fast heart beat, fever, shortness of breath, feeling faint or light-headed within 24 hours of
infusion.
-
Blockage in the gut or bowel
-
Bleeding in the stomach, shown by the vomiting of blood or the passage of red or black stools
-
Inflammation and pain or burning and stinging in the area around the catheter
-
Infection of the skin
-
Fits
-
Lung problems including blood clot in the lungs or accumulation of fluid around the lungs
which cause chest pain and breathlessness
-
Severe skin reactions such as flaking of the skin, rash and sensitive skin
-
Severe kidney problems
Other side effects
Very common side effects:
-
Feeling sick (nausea), vomiting and diarrhoea
-
Tiredness, pain and chills
-
Reduction in number of white blood cells
Common side effects:
-
Increased number of white blood cells
-
Increased clotting factors
-
Reduction in red blood cells (anaemia)
-
Decreased blood levels of calcium, potassium and sodium
-
Decreased blood protein levels
-
High blood sugar
-
A very fast heart beat
-
Spinning sensation
-
Constipation, indigestion, stomach problems, heartburn, passing wind and mouth ulcers
-
Flu-like symptoms
-
Fluid retention
-
Dizziness
or headache
-
Chest pain
-
Increased sweating, feeling thirsty and weak
-
Liver problems and yellowing of the skin (jaundice)
-
Infections
including bladder infections
-
Lumps under the skin on the back of the legs that may become sores and leave scars
-
Increased protein levels or white blood cells in urine
-
Loss of appetite
-
Dehydration
-
Back pain, aching muscles and joints
-
Feeling anxious and having difficulty sleeping.
-
Passing small amounts of urine or finding blood in the urine
-
Skin redness, itchy rash, hives, sensitive skin or a sudden widespread rash
-
High or low blood pressure
-
Flushing and hot flushes.
If any of the side effects become serious or if you notice any side effects not listed in this leaflet, tell
your doctor or nurse.
5.
HOW TO STORE REMOVAB
40
Keep out of the reach and sight of children.
Do not use Removab after the expiry date which is stated on the carton after EXP. The expiry date
refers to the last day of that month.
Store in a refrigerator (2°C – 8°C). Do not freeze. Store in the original package in order to protect
from light.
The prepared infusion solution should be used immediately.
6.
FURTHER INFORMATION
What Removab contains
-
The active substance is catumaxomab (10 microgram in 0.1 ml, corresponding to 0.1 mg/ml).
-
The other ingredients are sodium citrate, citric acid monohydrate, polysorbate 80 and water for
injections.
What Removab looks like and contents of the pack
Removab is presented as a clear and colourless concentrate for solution for infusion in a pre-filled
syringe with a cannula. Pack size of 1.
Marketing Authorisation Holder and Manufacturer
Fresenius Biotech GmbH
Am Haag 6-7
82166 Graefelfing
Germany
For any information about this medicine, please contact the Marketing Authorisation Holder.
This leaflet was last approved in
MM/YYYY.
Detailed information on this medicine is available on the European Medicines Agency (EMEA) web
site:
http://www.emea.europa.eu/
.
---------------------------------------------------------------------------------------------------------------------------
--
The following information is intended for medical or healthcare professionals only:
For information on dilution and administration of Removab please refer to section 6.6 of the Summary
of Product Characteristics (SPC) included in each package of Removab 10 microgram and Removab
50 microgram, respectively.
41
PACKAGE LEAFLET: INFORMATION FOR THE USER
Removab 50 microgram concentrate for solution for infusion
catumaxomab
Read all of this leaflet carefully before you start using this medicine.
-
Keep this leaflet. You may need to read it again.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
-
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor.
In this leaflet:
1.
What Removab is and what it is used for
2.
Before you use Removab
3.
How to use Removab
4.
How to store Removab
6.
Further information
1.
WHAT REMOVAB IS AND WHAT IT IS USED FOR
Removab contains the active substance catumaxomab, a monoclonal antibody. It recognises a protein
on the surface of cancer cells and recruits immune cells to destroy them.
Removab is used to treat malignant ascites,
when standard treatment is not available or no longer
feasible. Malignant ascites is an accumulation of fluid in the abdominal space (peritoneal cavity)
resulting from certain types of cancer.
2.
BEFORE YOU USE REMOVAB
Do not use Removab
-
if you are allergic (hypersensitive) to catumaxomab or any of the other ingredients of Removab
(see section 6)
-
if you are allergic (hypersensitive) to murine proteins (from rat and / or mouse)
Take special care with Removab
It is important to tell your doctor
if you have any of the following.
-
undrained fluid in your abdominal cavity
-
cold hands and feet, light headedness, difficulty passing urine, increased heart rate, and
weakness(symptoms of low blood volume)
-
weight gain, weakness, shortness of breath and fluid retention (symptoms of low blood protein
levels)
-
feeling dizzy and faint (symptoms of low blood pressure)
-
problems with your heart and circulation.
-
kidney or liver problems
Removab should not be used in children and adolescents under 18 years of age.
Before you start using Removab your doctor will check your:
-
Body Mass Index (BMI), which depends on your height and weight
42
-
If you have any further questions, ask your doctor.
5.
Possible side effects
-
an infection.
- Karnofsky Index, a measure of your general performance status.
You are required to have a BMI above 17 (after drainage of ascites fluid) and a Karnofsky Index
above 60 to use this medicine.
Taking other medicines
Please tell your doctor or pharmacist if you are taking or have recently taken any other medicines
including those obtained without a prescription.
Pregnancy and breast-feeding
You should not use Removab if you are pregnant unless clearly necessary. Talk to your doctor if you
are, might be or are planning to become pregnant.
If you are breast-feeding, talk to your doctor before starting treatment.
Driving and using machines
There are no studies on the effects of Removab on the ability to drive and use machines. However, if
you experience side effects such as dizziness or chills during or after administration, you should not
drive or use machines until they disappear.
3.
HOW TO USE REMOVAB
You will be given Removab under the supervision of a doctor experienced in treating cancer. After the
Removab infusion you will be observed as decided by your doctor.
Before starting and during treatment, you will be given other medicines to reduce fever, pain or
inflammation caused by Removab.
A catheter will be placed in your abdominal space (intraperitoneal) for the whole treatment period,
until the day after your last infusion.
Removab is given as 4 intraperitoneal infusions with increasing dose (10, 20, 50 and 150 micrograms),
separated at least by a 2-day break.
If you have any further questions on the use of this product, ask your doctor.
4.
POSSIBLE SIDE EFFECTS
Like all medicines, Removab can cause side effects, although not everybody gets them.
These side effects may occur with certain frequencies, which are defined as follows:
very common: affects more than 1 user in 10
common: affects 1 to 10 users in 100
uncommon: affects 1 to 10 users in 1,000
rare: affects 1 to 10 users in 10,000
very rare: affects less than 1 user in 10,000
not known: frequency cannot be estimated from the available data.
Serious side effects
Some of these side effects may be serious and require medical treatment. You should tell a doctor
immediately if you experience any of these serious side effects.
Very common serious side effects:
-
Abdominal pain
-
Fever
43
Common serious side effects:
-
Abdominal pain accompanied by difficulty passing stools
-
Shortness of breath
Uncommon serious side effects:
-
Very fast heart beat, fever, shortness of breath, feeling faint or light-headed within 24 hours of
infusion.
-
Blockage in the gut or bowel
-
Bleeding in the stomach, shown by the vomiting of blood or the passage of red or black stools
-
Inflammation and pain or burning and stinging in the area around the catheter
-
Infection of the skin
-
Fits
-
Lung problems including blood clot in the lungs or accumulation of fluid around the lungs
which cause chest pain and breathlessness
-
Severe skin reactions such as flaking of the skin, rash and sensitive skin
-
Severe kidney problems
Other side effects
Very common side effects:
-
Feeling sick (nausea), vomiting and diarrhoea
-
Tiredness, pain and chills
-
Reduction in number of white blood cells
Common side effects:
-
Increased number of white blood cells
-
Increased clotting factors
-
Reduction in red blood cells (anaemia)
-
Decreased blood levels of calcium, potassium and sodium
-
Decreased blood protein levels
-
High blood sugar
-
A very fast heart beat
-
Spinning sensation
-
Constipation, indigestion, stomach problems, heartburn, passing wind and mouth ulcers
-
Flu-like symptoms
-
Fluid retention
-
Dizziness
or headache
-
Chest pain
-
Increased sweating, feeling thirsty and weak
-
Liver problems and yellowing of the skin (jaundice)
-
Infections
including bladder infections
-
Lumps under the skin on the back of the legs that may become sores and leave scars
-
Increased protein levels or white blood cells in urine
-
Loss of appetite
-
Dehydration
-
Back pain, aching muscles and joints
-
Feeling anxious and having difficulty sleeping.
-
Passing small amounts of urine or finding blood in the urine
-
Skin redness, itchy rash, hives, sensitive skin or a sudden widespread rash
-
High or low blood pressure
-
Flushing and hot flushes.
If any of the side effects become serious or if you notice any side effects not listed in this leaflet, tell
your doctor or nurse.
5.
HOW TO STORE REMOVAB
44
Keep out of the reach and sight of children.
Do not use Removab after the expiry date which is stated on the carton after EXP. The expiry date
refers to the last day of that month.
Store in a refrigerator (2°C – 8°C). Do not freeze. Store in the original package in order to protect
from light.
The prepared infusion solution should be used immediately.
6.
FURTHER INFORMATION
What Removab contains
-
The active substance is catumaxomab (50 microgram in 0.5 ml, corresponding to 0.1 mg/ml).
-
The other ingredients are sodium citrate, citric acid monohydrate, polysorbate 80 and water for
injections.
What Removab looks like and contents of the pack
Removab is presented as a clear and colourless concentrate for solution for infusion in a pre-filled
syringe with a cannula. Pack size of 1.
Marketing Authorisation Holder and Manufacturer
Fresenius Biotech GmbH
Am Haag 6-7
82166 Graefelfing
Germany
For any information about this medicine, please contact the Marketing Authorisation Holder.
This leaflet was last approved in
MM/YYYY.
Detailed information on this medicine is available on the European Medicines Agency (EMEA) web
site:
http://www.emea.europa.eu/
.
---------------------------------------------------------------------------------------------------------------------------
--
The following information is intended for medical or healthcare professionals only:
For information on dilution and administration of Removab please refer to section 6.6 of the Summary
of Product Characteristics (SPC) included in each package of Removab 10 microgram and Removab
50 microgram, respectively.
45
Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/removab.html
Copyright © 1995-2021 ITA all rights reserved.