










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Retacrit 1000 IU/0.3ml solution for injection in pre-filled syringe
2.
1 pre-filled syringe with 0.3 ml solution for injection contains 1000 international units (IU) epoetin
zeta* (recombinant human erythropoietin). The solution contains 3333 IU Epoetin zeta per ml.
*Produced by recombinant DNA technology in Chinese Hamster Ovary (CHO) cell line.
Excipient:
Each pre-filled syringe contains 0.15 mg phenylalanine.
For a full list of excipients, see section 6.1.
3.
Solution for injection in pre-filled syringe.
Clear, colourless solution.
4.
4.1
−
Treatment of symptomatic anaemia associated with chronic renal failure (CRF) in adult and
paediatric patients:
o
Treatment of anaemia associated with chronic renal failure in adult and paediatric
patients on haemodialysis and adult patients on peritoneal dialysis (See section 4.4).
o
Treatment of severe anaemia of renal origin accompanied by clinical symptoms in
adult patients with renal insufficiency not yet undergoing dialysis (See section 4.4).
−
Treatment of anaemia and reduction of transfusion requirements in adult patients receiving
chemotherapy for solid tumours, malignant lymphoma or multiple myeloma, and at risk of
transfusion as assessed by the patient's general status (e.g. cardiovascular status, pre-existing
anaemia at the start of chemotherapy).
−
Retacrit can be used to increase the yield of autologous blood from patients in a predonation
programme. Its use in this indication must be balanced against the reported risk of
thromboembolic events. Treatment should only be given to patients with moderate anaemia (no
iron deficiency), if blood saving procedures are not available or insufficient when the scheduled
major elective surgery requires a large volume of blood (4 or more units of blood for females or
5 or more units for males).
4.2
Treatment with Retacrit has to be initiated under the supervision of physicians experienced in the
management of patients with above indications.
Posology
Treatment of symptomatic anaemia in adult and paediatric chronic renal failure patients
Retacrit should be administered either subcutaneously or intravenously.
2
The haemoglobin concentration aimed for is between 10 and 12 g/dl (6.2-7.5 mmol/l), except in
paediatric patients in whom the haemoglobin concentration should be between 9.5 and 11 g/dl
(5.9-6.8 mmol/l). The upper limit of the target haemoglobin concentration should not be exceeded.
Anemia symptoms and sequaelea may vary with age, gender and overall burden of disesase; a
physician´s evaluation of the individual patient´s clinical course and condition is necessary. Retacrit
should be administered either subcutaneously or intravenously in order to increase haemoglobin to not
greater than 12 g/dL (7.5 mmol/L) Due to intra-patient variability, occasional individual haemoglobin
values for a patient above and below the desired haemoglobin level may be observed. Haemoglobin
variability should be addressed through dose management, with consideration for the haemoglobin
target range of 10 g/dL (6.2 mmol/l) to 12 g/dl (7.5 mmol/l).
A sustained haemoglobin level of greater than 12 g/dl should be avoided; guidance for appropriate
dose adjustment for when haemoglobin values exceeding 12 g/dl (7.5 mmol/l) are observed are
described below. A rise in haemoglobin of greater than 2 g/dL (1.25 mmol/l) over a four week period
should be avoided. If it occurs, appropriate dose adjustment should be made as provided.
Patients should be monitored closely to ensure that the lowest approved dose of Retacrit is used to
provide adequate control of the symptoms of anemia.
In patients with chronic renal failure and clinically evident ischemic heart disease or congestive heart
failure, maintenance haemoglobin concentration should not exceed the upper limit of the target
haemoglobin concentration.
Adult patients on haemodialysis
Retacrit should be administered either subcutaneously or intravenously.
The treatment is divided into two stages:
1. Correction phase: 50 IU/kg 3 times per week. When a dose adjustment is necessary, this should
be done in steps of at least four weeks. At each step, the increase or reduction
in dose should be of 25 IU/kg 3 times per week.
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 10 and 12 g/dl (6.2-7.5 mmol/l). The recommended total
weekly dose is between 75 and 300 IU/kg.
The clinical data available suggest that those patients whose initial haemoglobin is very low (< 6 g/dl
or < 3.75 mmol/l) may require higher maintenance doses than those whose initial anaemia is less
severe (Hb > 8 g/dl or > 5 mmol/l).
Paediatric patients on haemodialysis
1. Correction phase 50 IU/kg, 3 times per week by the intravenous route. When a dose adjustment
is necessary, this should be done in steps of 25 IU/kg, 3 times per week at
intervals of at least 4 weeks until the desired goal is achieved.
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 9.5 and 11 g/dl (5.9-6.8 mmol/l).
Generally, children and adolescents under 30 kg body weight require higher maintenance doses than
adults and children over 30 kg. The following maintenance doses were observed in clinical trials after
6 months of treatment.
3
The treatment is divided into two stages:
Dose (IU/kg given 3x week)
Weight (kg)
Median
Usual maintenance dose
< 10
100
75-150
10-30
75
60-150
> 30
33
30-100
The clinical data available suggest that those patients whose initial haemoglobin is very low
(< 6.8 g/dl or < 4.25 mmol/l) may require higher maintenance doses than those whose initial
haemoglobin is higher > 6.8 g/dl or> 4.25 mmol/l).
Adult patients on peritoneal dialysis
Retacrit should be administered either subcutaneously or intravenously.
The treatment is divided into two stages:
1. Correction phase: Starting dose of 50 IU/kg 2 times per week.
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 10 and 12 g/dl (6.2-7.5 mmol/l). Maintenance dose between
25 and 50 IU/kg 2 times per week into 2 equal doses.
Adult patients with renal insufficiency not yet undergoing dialysis
Retacrit should be administered either subcutaneously or intravenously.
The treatment is divided into two stages:
1. Correction phase: Starting dose of 50 IU/kg 3 times per week, followed if necessary by a dose
increase with 25 IU/kg increments (3 times per week) until the desired goal is
achieved (this should be done in steps of at least four weeks).
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 10 and 12 g/dl (6.2-7.5 mmol/l). Maintenance dose between
17 and 33 IU/kg 3 times per week.
The maximum dose should not exceed 200 IU/kg 3 times per week.
−
Treatment of patients with chemotherapy induced anaemia..
Retacrit should be administered by the subcutaneous route to patients with anaemia (e.g. haemoglobin
concentration ≤ 10 g/dl (6.2 mmol/l). Anaemia symptoms and sequelae may vary with age, gender,
and overall burden of disease; a physician´s evaluation of the individual patient´s clinical course and
condition is necessary.
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management with consideration for the haemoglobin target range of 10 g/dl (6.2 mmol/l)
to 12 g/dl (7.5 mmol/l). A sustained haemoglobin level of greater than 12 g/dl (7.5 mmol/l) should be
avoided; guidance for appropriate dose adjustment for when haemoglobin values exceeding 12 g/dl
(7.5 mmol/l) are observed are described below.
Patients should be monitored closely to ensure that the lowest approved dose of Retacrit is used to
provide adequate control of the symptoms of anaemia.
Retacrit therapy should continue until one month after the end of chemotherapy.
4










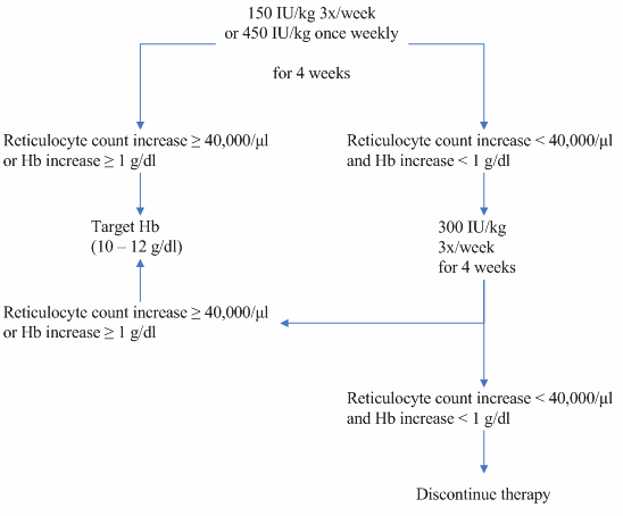
The initial dose is 150 IU/kg given subcutaneously 3 times per week. Alternatively, Retacrit can be
administered at an initial dose of 450 IU/kg subcutaneously once weekly.
If the haemoglobin has increased by at least 1 g/dl (0.62 mmol/l) or the reticulocyte count has
increased ≥ 40,000 cells/µl above baseline after 4 weeks of treatment, the dose should remain at
150 IU/kg 3 times per week or 450 IU/kg once weekly. If the haemoglobin increase is < 1 g/dl
(< 0.62 mmol/l) and the reticulocyte count has increased < 40,000 cells/µl above baseline, increase the
dose to 300 IU/kg 3 times per week. If after an additional 4 weeks of therapy at 300 IU/kg 3 times per
week, the haemoglobin has increased ≥ 1 g/dl (0.62 mmol/l) or the reticulocyte count has increased
≥ 40,000 cells/µl the dose should remain at 300 IU/kg 3 times per week. However, if the haemoglobin
has increased < 1 g/dl (< 0.62 mmol/l) and the reticulocyte count has increased < 40,000 cells/µl above
baseline, response is unlikely and treatment should be discontinued.
The recommended dosing regimen is described in the following diagram:
Once the therapeutic objective for an individual patient has been achieved, the dose should be reduced
by 25 to 50% in order to maintain haemoglobin at that level. Appropriate dose titration should be
considered.
Dose adjustment
At a rate of rise in haemoglobin of > 2 g/dl (> 1.25 mmol/l) per month the Retacrit dose should be
reduced by about 25-50%. If haemoglobin level exceeds 12 g/dl (7.5 mmol/l), discontinue therapy
until it falls to 12 g/dl (7.5 mmol/l) or lower and then reinstitute Retacrit therapy at a dose 25% below
the previous dose.
−
Adult surgery patients in an autologous predonation programme.
5
Retacrit should be given by the intravenous route.
At the time of donating blood, Retacrit should be administered after the completion of the blood
donation procedure.
Mildly anaemic patients (haematocrit of 33-39%) requiring predeposit of ≥ 4 units of blood should be
treated with Retacrit at a dose of 600 IU/kg body weight 2 times weekly for 3 weeks prior to surgery.
All patients being treated with Retacrit should receive adequate iron supplementation (e.g. 200 mg oral
elemental iron daily) throughout the course of treatment. Iron supplementation should be started as
soon as possible, even several weeks prior to initiating the autologous predeposit, in order to achieve
high iron stores prior to starting Retacrit therapy.
Method of administration
For instructions on handling of the medicinal product before administration, see section 6.6.
The dose should be administered over at least 1-5 minutes, depending on the total dose. In
haemodialysed patients, a bolus injection may be given during the dialysis session through a suitable
venous port in the dialysis line. Alternatively, the injection can be given at the end of the dialysis
session via the fistula needle tubing, followed by 10 ml of sodium chloride 9 mg/ml (0.9%) solution
for injection to rinse the tubing and ensure satisfactory injection of the medicinal product into the
circulation.
A slower injection is preferable in patients who react to the treatment with “flu-like” symptoms.
Retacrit should not be administered by intravenous infusion.
Retacrit must not be mixed with other medicinal products (see section 6.2).
Subcutaneous injection
A maximum volume of 1 ml at one injection site should generally not be exceeded. In case of larger
volumes, more than one site should be chosen for the injection.
The injections are given in the limbs or the anterior abdominal wall.
4.3
−
Hypersensitivity to the active substance or to any of the excipients.
−
Patients who develop Pure Red Cell Aplasia (PRCA) following treatment with any
erythropoietin must not receive Retacrit or any other erythropoietin (see section 4.4).
−
Uncontrolled hypertension.
−
In the indication "increasing the yield of autologous blood": myocardial infarction or stroke in
the month preceding treatment, unstable angina pectoris, increased risk of deep venous
thrombosis such as history of venous thromboembolic disease.
−
Patients who for any reason cannot receive adequate antithrombotic prophylaxis.
4.4
General
Like in all patients receiving erythropoietin, blood pressure may rise during treatment with Retacrit.
Blood pressure should be closely monitored and adequately controlled in all epoetin treatment naïve as
well as pre-treated patients before, at initiation of, and during treatment with Retacrit. It may be
necessary to add or increase anti-hypertensive treatment. If blood pressure cannot be well controlled,
Retacrit treatment should be discontinued.
6
Intravenous injection
Retacrit should also be used with caution in the presence of epilepsy and chronic liver failure.
There may be a moderate dose-dependent rise in the platelet count within the normal range during
treatment with erythropoietin. This regresses during the course of continued therapy. It is
recommended that the platelet count is regularly monitored during the first 8 weeks of therapy.
All other causes of anaemia (iron deficiency, haemolysis, blood loss, vitamin B
12
- or folate
deficiencies) should be considered and treated prior to initiating and during therapy with Retacrit. In
most cases, the ferritin values in the serum fall simultaneously with the rise in packed cell volume. In
order to ensure optimum response to erythropoietin, adequate iron stores should be assured:
−
iron supplementation, e.g. 200-300 mg/day orally (100-200 mg/day for paediatric patients) is
recommended for chronic renal failure patients whose serum ferritin levels are below 100 ng/ml
−
oral iron substitution of 200-300 mg/day is recommended for all cancer patients whose
transferrin saturation is below 20%.
All of these additive factors of anaemia should also be carefully considered when deciding to increase
the dose of erythropoietin in cancer patients.
A paradoxical decrease in haemoglobin and development of severe anaemia associated with low
reticulocyte counts should prompt to discontinue treatment with epoetin and perform anti-
erythropoietin antibody testing. Cases have been reported in patients with hepatits C treated with
interferon and ribavirin, when epoetins are used concomitantly. Epoetins are not approved in the
management of anaemia associated with hepatitis C.
In order to improve the traceability of ESAs, the name of the prescribed ESA should be clearly
recorded (or: stated) in the patient file.
Good blood management practices should always be used in the perisurgical setting.
This medicinal product contains phenylalanine which may be harmful for people with
phenylketonuria.
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, i.e. essentially
‘sodium-free’.
Chronic renal failure patients
Haemoglobin concentration
In patients with chronic renal failure, maintenance haemoglobin concentration should not exceed the
upper limit of the target haemoglobin concentration recommended in section 4.2. In clinical trials, an
increased risk of death, serious cardiovascular events or cerebrovascular events including stroke were
observed when ESAs were administered to target a haemoglobin of greater than 12 g/dl (7.5 mmol/l).
Controlled clinical trials have not shown significant benefits attributable to the administration of
epoetins when haemoglobin concentration is increased beyond the level necessary to control
symptoms of anaemia and to avoid blood transfusion.
Haemoglobin levels should be measured on a regular basis until a stable level is achieved and
periodically thereafter. The rate of increase in haemoglobin should be approximately 1 g/dl
(0.62 mmol/l) per month and should not exceed 2 g/dl (1.25 mmol/l) per month to minimize the risk of
developing or worsening of hypertension.
Chronic renal failure patients treated with Retacrit by the subcutaneous route should be monitored
regularly for loss of efficacy, defined as absent or decreased response to Retacrit treatment in patients
who previously responded to such therapy. This is characterised by a sustained decrease in
haemoglobin despite an increase in Retacrit dosage.
7
Non response to erythropoietin therapy should prompt a search for causative factors. These include:
iron, folate, or Vitamin B
12
deficiency; aluminium intoxication; intercurrent infections; inflammatory
or traumatic episodes; occult blood loss; haemolysis, and bone marrow fibrosis of any origin.
Cases of antibody-mediated PRCA have been very rarely reported in chronic renal failure patients
with erythropoietin administered by the subcutaneous route. In patients developing sudden lack of
efficacy, defined by a decrease in haemoglobin (1-2 g/dl per month) with increased need for
transfusions, a reticulocyte count should be obtained and typical causes of non-response (e.g. iron,
folate, or Vitamin B
12
-deficiency, aluminium intoxication, infection or inflammation, blood loss, and
haemolysis) should be investigated. If no cause is identified, a bone marrow examination should be
considered for diagnosis of PRCA.
If PRCA is diagnosed, therapy with Retacrit must be immediately discontinued and testing for
erythropoietin antibodies should be considered. Patients should not be switched to another medicinal
product as anti-erythropoietin antibodies cross-react with other erythropoietins. Other causes of PRCA
should be excluded, and appropriate therapy initiated.
Monitoring of reticulocyte count on a regular basis is recommended to detect possible occurrence of
lack of efficacy in chronic renal failure patients.
Hyperkalaemia has been observed in isolated cases. In chronic renal failure patients, correction for
anaemia may lead to increased appetite, and potassium and protein intake. Dialysis prescriptions may
have to be adjusted periodically to maintain urea, creatinine and potassium in the desired range. Serum
electrolytes should be monitored in chronic renal failure patients. If an elevated (or rising) serum
potassium level is detected then consideration should be given to ceasing erythropoietin administration
until hyperkalaemia has been corrected.
An increase in heparin dose during haemodialysis is frequently required during the course of therapy
with erythropoietin as a result of the increased packed cell volume. Occlusion of the dialysis system is
possible if heparinisation is not optimum.
Based on information available to date, correction of anaemia with erythropoietin in adult patients
with renal insufficiency not yet undergoing dialysis does not accelerate the rate of progression of renal
insufficiency.
Adult cancer patients with symptomatic anaemia receiving chemotherapy
In cancer patients receiving chemotherapy, the 2-3 week delay between erythropoietin administration
and the appearance of erythropoietin-induced red cells should be taken into account when assessing if
Retacrit therapy is appropriate (patient at risk of being transfused).
Haemoglobin levels should be closely monitored until a stable level is achieved and periodically
thereafter. If the rate of increase in haemoglobin exceeds 2 g/dl (1.25 mmol/l) per month or the
haemoglobin level exceeds 12 g/dl (7,5 mmol/l), the dose adjustment detailed in section 4.2 should be
thoroughly performed to minimise the risk of thrombotic events (see section 4.2).
As an increased incidence of thrombotic vascular events (TVEs) has been observed in cancer patients
receiving erythropoietic agents (see section 4.8), this risk should be carefully weighed against the
benefit to be derived from treatment (with Retacrit) particularly in cancer patients with an increased
risk of thrombotic vascular events, such as obesity and patients with a prior history of TVEs (e.g. deep
venous thrombosis or pulmonary embolism).
Adult surgery patients in an autologous predonation programme
All special warnings and precautions associated with autologous predonation programs, especially
routine volume replacement, should be respected.
8
Tumour growth potential
Epoetins are growth factors that primarily stimulate red blood cell production. Erythropoietin
receptors may be expressed on the surface of a variety of tumour cells. As with all growth factors,
there is a concern that epoetins could stimulate the growth of any type of malignancy. In several
controlled studies, epoetins have not been shown to improve overall survival or decrease the risk of
tumour progression in patients with anaemia associated with cancer.
Several controlled clinical studies in which epoetins were administered to patients with a variety of
common tumours including squamous head and neck cancer, lung cancer, and breast cancer, have
shown an unexplained excess mortality.
In controlled clinical studies, use of Epoetin alfa and other erythropoiesis-stimulating agents (ESAs)
have shown:
•
shortened time to tumour progression in patients with advanced head and neck cancer
receiving radiation therapy when administered to target a haemoglobin of greater than 14 g/dl
(8.7 mmol/l),
•
shortened overall survival and increased deaths attributed to disease progression at 4 months
in patients with metastatic breast cancer receiving chemotherapy when administered to target a
haemoglobin of 12-14 g/dl (7.5 -8.7 mmol/l),
•
increased risk of death when administered to target a haemoglobin of 12 g/dl (7.5 mmol/l) in
patients with active malignant disease receiving neither chemotherapy nor radiation therapy.
ESAs are not indicated for use in this patient population.
In view of the above, in some clinical situations blood transfusion should be the preferred treatment
for the management of anaemia in patients with cancer. The decision to administer recombinant
erythropoietins should be based on a benefit-risk assessment with the participation of the individual
patient, which should take into account the specific clinical context. Factors that should be considered
in this assessment should include the type of tumour and its stage; the degree of anaemia; life-
expectancy; the environment in which the patient is being treated; and patient preference (see section
5.1).
4.5
There is no evidence to indicate that treatment with erythropoietin alters the metabolism of other
medicinal products.
However, since ciclosporin is bound by red blood cells there is potential for interactions with other
medicinal products. If erythropoietin is given concomitantly with ciclosporin, blood levels of
ciclosporin should be monitored and the dose of ciclosporin adjusted as the haematocrit rises.
No evidence exists that indicates an interaction between epoetin alfa and G-CSF or GM-CSF with
regard to haematological differentiation or proliferation of tumour biopsy specimens in vitro.
4.6
There are no adequate and well-controlled studies in pregnant women. Studies in animals have shown
reproduction toxicity (see section 5.3). Consequently, erythropoietin should generally be used during
pregnancy and lactation only if the potential benefit outweighs the potential risk to the foetus.
4.7
Retacrit has no or negligible influence on the ability to drive and use machines.
4.8
9
Retacrit is a biological medicinal product. Data from clinical studies with Retacrit are in line with the
safety profile of other authorized erythropoietins. Based on the results from clinical trials with other
authorized erythropoietins approximately 8% of patients treated with erythropoietin are expected to
predominantly in patients with chronic renal failure or underlying malignancies. These undesirable
effects are most commonly headache and a dose dependent increase in blood pressure. Hypertensive
crisis with encephalopathy-like symptoms can occur. Attention should be paid to sudden stabbing
migraine-like headaches as a possible warning signal.
Thrombotic/vascular events, such as myocardial ischaemia, myocardial infarction, cerebrovascular
accidents (cerebral haemorrhage and cerebral infarction), transient ischaemic attacks, deep vein
thrombosis, arterial thrombosis, pulmonary emboli, aneurysms, retinal thrombosis, and clotting of an
artificial kidney have been reported in patients receiving erythropoietic agents.
Antibody-mediated erythroblastopenia (PRCA) has been reported after months to years of treatment
with epoetin alfa. In most of these patients, antibodies to erythropoietins have been observed (see
sections 4.3 and 4.4).
In this section frequencies of undesirable effects are defined as follows: Very common (
>
1/10);
common (
>
1/100 to <1/10); uncommon (
>
1/1,000 to <1/100); rare (
>
1/10,000 to <1/1,000); very rare
(<1/10,000), not known (frequency cannot be estimated from the available data).
SOC
Frequency
ADR
very rare
Thrombocytosis (see section 4.4)
Blood and lymphatic system
disorders
Frequency not known
Antibody-mediated erythroblastopenia
(PRCA)
Immune system disorders
rare
Hypersensitivity reactions
very rare
anaphylactic reaction
very common
dizziness (chronic renal failure
patients)
headache (cancer patients)
Nervous system disorders
common
dizziness (cancer patients)
headache (chronic renal failure
patients)
stroke
uncommon
cerebral haemorrhage
Frequency not known
cerebral infarction
transient ischaemic attacks
hypertensive encephalopathy
Eye disorders
Frequency not known
retinal thrombosis
Cardiac disorders
Frequency not known
myocardial infarction
myocardial ischaemia
common
deep vein thrombosis (cancer patients)
increase in blood pressure
Vascular disorders
aneurysms
arterial thrombosis
deep vein thrombosis (chronic renal
failure patients)
hypertensive crisis
Frequency not known
Respiratory, thoracic and
mediastinal disorders
common
pulmonary embolism (cancer patients)
Frequency not known
pulmonary embolism (chronic renal
failure patients)
Skin and subcutaneous tissue
disorders
common
Non-specific skin rashes
very rare
Angioedema
10












































Frequency not known
pruritus
very common
joint pains (chronic renal failure
patients)
Musculoskeletal and
connective tissue disorders
common
joint pains (cancer patients)
very common
"Flu-like" symptoms (chronic renal
failure patients)
feelings of weakness (chronic renal
failure patients)
tiredness (chronic renal failure
patients)
General disorders and
administration site conditions
common
"Flu-like" symptoms (cancer patients)
feelings of weakness (cancer patients)
tiredness (cancer patients)
Injury, poisoning and
procedural complications
common
clotting of an artificial kidney
Adult and paediatric haemodialysis patients, adult peritoneal dialysis patients and adult patients with
renal insufficiency not yet undergoing dialysis
The most frequent adverse reaction during treatment with epoetin alfa is a dose-dependent increase in
blood pressure or aggravation of existing hypertension. These increases in blood pressure can be
treated with medicinal products. Moreover, monitoring of the blood pressure is recommended
particularly at the start of therapy. The following reactions have also occurred in isolated patients with
normal or low blood pressure: hypertensive crisis with encephalopathy-like symptoms (e.g. headaches
and confused state) and generalised tonoclonal seizures, requiring the immediate attention of a
physician and intensive medical care. Particular attention should be paid to sudden stabbing migraine
like headaches as a possible warning signal.
Shunt thromboses may occur, especially in patients who have a tendency to hypotension or whose
arteriovenous fistulae exhibit complications (e.g. stenoses, aneurysms, etc.). Early shunt revision and
thrombosis prophylaxis by administration of acetylsalicylic acid, for example, is recommended in
these patients.
Adult cancer patients with symptomatic anaemia receiving chemotherapy
Hypertension may occur in epoetin alfa treated patients. Consequently, haemoglobin and blood
pressure should be closely monitored.
An increased incidence of thrombotic vascular events (see section 4.4 and section 4.8 - General) has
been observed in patients receiving erythropoietic agents.
Surgery patients in autologous predonation programmes
Independent of erythropoietin treatment, thrombotic and vascular events may occur in surgical patients
with underlying cardiovascular disease following repeated phlebotomy. Therefore, routine volume
replacement should be performed in such patients.
4.9
The therapeutic margin of erythropoietin is very wide. Overdose of erythropoietin may produce effects
that are extensions of the pharmacological effects of the hormone. Phlebotomy may be performed if
excessively high haemoglobin levels occur. Additional supportive care should be provided as
necessary.
5.
11




















5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Antianaemic preparations, erythropoietin
ATC code: B03XA01
Retacrit is a biosimilar medicinal product. Detailed information is available on the website of the
European Medicines Agency http://www.ema.europa.eu.
Erythropoietin is a glycoprotein that stimulates, as a mitosis-stimulating factor and differentiating
hormone, the formation of erythrocytes from precursors of the stem cell compartment.
The apparent molecular weight of erythropoietin is 32,000-40,000 Dalton. The protein moiety of the
molecule contributes about 58% of total molecular weight and consists of 165 amino acids. The four
carbohydrate chains are attached via three N-glycosidic bonds and one O-glycosidic bond to the
protein. Epoetin zeta is identical in its amino acid sequence and similar in carbohydrate composition to
endogenous human erythropoietin that has been isolated from the urine of anaemic patients.
The biological efficacy of erythropoietin has been demonstrated in various animal models
in vivo
(normal and anaemic rats, polycythaemic mice). After administration of erythropoietin, the number of
erythrocytes, the Hb values and reticulocyte counts increase as well as the
59
Fe-incorporation rate.
An increased
3
H-thymidine incorporation in the erythroid nucleated spleen cells has been found
in vitro
(mouse spleen cell culture) after incubation with erythropoietin. It could be shown with the aid
of cell cultures of human bone marrow cells that erythropoietin stimulates erythropoiesis specifically
and does not affect leucopoiesis. Cytotoxic actions of erythropoietin on bone marrow cells could not
be detected.
As with other haematopoietic growth factors, erythropoietin has shown
in vitro
stimulating properties
on human endothelial cells.
721 cancer patients receiving non-platinum chemotherapy were included in three placebo-controlled
studies, 389 patients with haematological malignancies (221 multiple myeloma, 144 non-Hodgkin's
lymphoma, and 24 other haematological malignancies) and 332 with solid tumours (172 breast,
64 gynaecological, 23 lung, 22 prostate, 21 gastrointestinal, and 30 other tumour types). In two large,
open-label studies, 2697 cancer patients receiving non-platinum chemotherapy were included,
1895 with solid tumours (683 breast, 260 lung, 174 gynaecological, 300 gastrointestinal, and 478 other
tumour types) and 802 with haematological malignancies.
In a prospective, randomised, double-blind, placebo-controlled trial conducted in 375 anaemic patients
with various non-myeloid malignancies receiving non-platinum chemotherapy, there was a significant
reduction of anaemia-related sequelae (e.g. fatigue, decreased energy, and activity reduction), as
measured by the following instruments and scales: Functional Assessment of Cancer
Therapy-Anaemia (FACT-An) general scale, FACT-An fatigue scale, and Cancer Linear Analogue
Scale (CLAS). Two other smaller, randomized, placebo-controlled trials failed to show a significant
improvement in quality of life parameters on the EORTC-QLQ-C30 scale or CLAS, respectively.
Erythropoietin is a growth factor that primarily stimulates red cell production. Erythropoietin receptors
may be expressed on the surface of a variety of tumour cells.
Survival and tumour progression have been examined in five large controlled studies involving a total
of 2833 patients, of which four were double-blind placebo-controlled studies and one was an open-
label study. The studies either recruited patients who were being treated with chemotherapy (two
studies) or used patient populations in which erythropoiesis stimulating agents are not indicated:
anaemia in patients with cancer not receiving chemotherapy, and head and neck cancer patients
receiving radiotherapy. The target haemoglobin concentration in two studies was > 13 g/dl; in the
remaining three studies it was 12-14 g/dl. In the open-label study there was no difference in overall
survival between patients treated with recombinant human erythropoietin and controls. In the four
12
placebo-controlled studies the hazard ratios for overall survival ranged between 1.25 and 2.47 in
favour of controls. These studies have shown a consistent unexplained statistically significant excess
mortality in patients who have anaemia associated with various common cancers who received
recombinant human erythropoietin compared to controls. Overall survival outcome in the trials could
not be statisfactorily explained by differences in the incidence of thrombosis and related complications
between those given recombinant human erythropoietin and those in the control group.
A systematic review has also been performed involving more than 9000 cancer patients participating
in 57 clinical trials. Meta-analysis of overall survival data produced a hazard ratio point estimate of
1.08 in favour of controls (95% CI: 0.99, 1,18; 42 trials and 8167 patients). An increased relative risk
of thromboembolic events (RR 1.67, 95% CI: 1.35, 2.06, 35 trials and 6769 patients) was observed in
patients treated with recombinant human erythropoietin. There is an increased risk for
thromboembolic events in patients with cancer treated with recombinant human erythropoietin and a
negative impact on overall survival cannot be excluded. The extent to which these outcomes might
apply to the administration of recombinant human erythropoietin to patients with cancer, treated with
chemotherapy to achieve haemoglobin concentrations less than 13 g/dl, is unclear because few patients
with these characteristics were included in the data reviewed.
A patient-level data analysis has also been performed on more than 13.900 cancer patients (chemo-,
radio-, chemoradio-, or no therapy) participating in 53 controlled clinical trials involving several
epoetins. Meta-analysis of overall survival data produced a hazard ratio point estimate of 1.06 in
favour of controls (95% CI: 1.00, 1.12; 53 trials and 13933 patients) and for the cancer patients
receiving chemotherapy, the overall survival hazard ratio was 1.04 (95% CI: 0.97, 1.11; 38 trials and
10.441 patients). Meta-analyses also indicate consistently a significantly increased relative risk of
thromboembolic events in cancer patients receiving recombinant human erythropoietin (see section
4.4).
In a randomised, double-blind, placebo-controlled study of 4,038 CRF patients not on dialysis with
type 2 diabetes and haemoglobin levels ≤ 11 g/dL, patients received either treatment with darbepoetin
alfa to target haemoglobin levels of 13 g/dL or placebo (see section 4.4). The study did not meet either
primary objective of demonstrating a reduction in risk for all-cause mortality, cardiovascular
morbidity, or end stage renal disease (ESRD). Analysis of the individual components of the composite
endpoints showed the following HR (95% CI): death 1.05 (0.92, 1.21), stroke 1.92 (1.38, 2.68),
congestive heart failure (CHF) 0.89 (0.74, 1.08), myocardial infarction (MI) 0.96 (0.75, 1.23),
hospitalisation for myocardial ischaemia 0.84 (0.55, 1.27), ESRD 1.02 (0.87, 1.18).
5.2
Pharmacokinetic properties
Measurement of erythropoietin following multiple dose intravenous administration revealed a half-life
of approximately 4 hours in healthy volunteers and a somewhat more prolonged half-life of
approximately 5 hours in renal failure patients. A half-life of approximately 6 hours has been reported
in children.
Following subcutaneous injection, serum levels of erythropoietin are much lower than the levels
achieved following intravenous injection, the levels increase slowly and reach a peak between 12 and
18 hours postdose. The peak is always well below the peak achieved using the intravenous route
(approximately 1/20th of the value).
There is no accumulation: the levels remain the same, whether they are determined 24 hours after the
first injection or 24 hours after the last injection.
The half-life is difficult to evaluate for the subcutaneous route and is estimated to be about 24 hours.
The bioavailability of subcutaneous injectable erythropoietin is much lower than that of the
intravenous medicinal product and is approximately 20%.
13
Intravenous route
Subcutaneous route
5.3
Preclinical safety data
In some pre-clinical toxicological studies in dogs and rats, but not in monkeys, erythropoietin therapy
was associated with subclinical bone marrow fibrosis (bone marrow fibrosis is a known complication
of chronic renal failure in humans and may be related to secondary hyperparathyroidism or unknown
factors. The incidence of bone marrow fibrosis was not increased in a study of haemodialysis patients
who were treated with erythropoietin for 3 years compared to a matched control group of dialysis
patients who had not been treated with erythropoietin).
In animal studies, erythropoietin has been shown to decrease foetal body weight, delay ossification
and increase foetal mortality when given in weekly doses of approximately 20 times the recommended
human weekly dose. These changes are interpreted as being secondary to decreased maternal body
weight gain.
Erythropoietin did not show any changes in bacterial and mammalian cell culture mutagenicity tests
and an
in vivo
micronucleus test in mice. Long-term carcinogenicity studies have not been carried out.
There are conflicting reports in the literature regarding whether erythropoietin may play a major role
as tumour proliferator. These reports are based on
in vitro
findings from human tumour samples, but
are of uncertain significance in the clinical situation.
6.
6.1
List of Excipients
Disodium phosphate dihydrate
Sodium dihydrogen phosphate dihydrate
Sodium chloride
Calcium chloride dihydrate
Polysorbate 20
Glycine
Leucine
Isoleucine
Threonine
Glutamic acid
Phenylalanine
Water for injections
Sodium hydroxide (pH adjuster)
Hydrochloric acid (pH adjuster)
6.2
Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3
Shelf life
2 years
6.4
Special precautions for storage
Store in a refrigerator (2°C - 8°C). Do not freeze.
Keep the pre-filled syringe in the outer carton in order to protect from light.
For the purpose of ambulatory use, the patient may remove the product from the refrigerator and store
it at room temperature (not above 25°C) for one single period of up to 3 days.
14
6.5
Nature and contents of container
Pre-filled syringe Type I glass with a fixed steel injection needle and a plunger stopper with PTFE
coating.
Each pre-filled syringe contains 0.3 ml solution for injection.
Each pack contains 1 or 6 pre-filled syringes.
Not all pack sizes may be marketed.
6.6
Special precautions for disposal and other handling
Handling instructions for Retacrit:
1.
After removing one syringe from the blister pack the solution should be checked to ensure that
it is clear, colourless and practically free from visible particles.
2.
The protective cap is removed from the injection needle and air is expelled from the syringe and
needle by holding the syringe vertically and gently pressing the plunger upwards.
3.
The syringe is now ready for use.
Retacrit must not be used if
•
The blister sealing is broken or the blister is damaged in any way.
•
The liquid is coloured or you can see particles floating in it.
•
Any liquid has leaked out of the pre-filled syringe or condensation is visible within the sealed
blister.
•
It may have been accidentally frozen.
This medicinal product is for single use only.
Do not shake.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Hospira UK Limited
Queensway
Royal Leamington Spa
Warwickshire
CV31 3RW
United Kingdom
8.
EU/1/07/431/001
EU/1/07/431/002
9.
18/12/2007
15
1.
Retacrit 2000 IU/0.6ml solution for injection in pre-filled syringe
2.
1 pre-filled syringe with 0.6 ml solution for injection contains 2000 international units (IU) epoetin
zeta* (recombinant human erythropoietin). The solution contains 3333 IU Epoetin zeta per ml.
*Produced by recombinant DNA technology in Chinese Hamster Ovary (CHO) cell line.
Excipient:
Each pre-filled syringe contains 0.30 mg phenylalanine.
For a full list of excipients, see section 6.1.
3.
Solution for injection in pre-filled syringe.
Clear, colourless solution.
4.
4.1
−
Treatment of symptomatic anaemia associated with chronic renal failure (CRF) in adult and
paediatric patients:
o
Treatment of anaemia associated with chronic renal failure in adult and paediatric
patients on haemodialysis and adult patients on peritoneal dialysis (See section 4.4).
o
Treatment of severe anaemia of renal origin accompanied by clinical symptoms in
adult patients with renal insufficiency not yet undergoing dialysis (See section 4.4).
−
Treatment of anaemia and reduction of transfusion requirements in adult patients receiving
chemotherapy for solid tumours, malignant lymphoma or multiple myeloma, and at risk of
transfusion as assessed by the patient's general status (e.g. cardiovascular status, pre-existing
anaemia at the start of chemotherapy).
−
Retacrit can be used to increase the yield of autologous blood from patients in a predonation
programme. Its use in this indication must be balanced against the reported risk of
thromboembolic events. Treatment should only be given to patients with moderate anaemia (no
iron deficiency), if blood saving procedures are not available or insufficient when the scheduled
major elective surgery requires a large volume of blood (4 or more units of blood for females or
5 or more units for males).
4.2
Treatment with Retacrit has to be initiated under the supervision of physicians experienced in the
management of patients with above indications.
Posology
Treatment of symptomatic anaemia in adult and paediatric chronic renal failure patients
Retacrit should be administered either subcutaneously or intravenously.
16
The haemoglobin concentration aimed for is between 10 and 12 g/dl (6.2-7.5 mmol/l), except in
paediatric patients in whom the haemoglobin concentration should be between 9.5 and 11 g/dl
(5.9-6.8 mmol/l). The upper limit of the target haemoglobin concentration should not be exceeded.
Anemia symptoms and sequaelea may vary with age, gender and overall burden of disesase; a
physician´s evaluation of the individual patient´s clinical course and condition is necessary. Retacrit
should be administered either subcutaneously or intravenously in order to increase haemoglobin to not
greater than 12 g/dL (7.5 mmol/L) Due to intra-patient variability, occasional individual haemoglobin
values for a patient above and below the desired haemoglobin level may be observed. Haemoglobin
variability should be addressed through dose management, with consideration for the haemoglobin
target range of 10 g/dL (6.2 mmol/l) to 12 g/dl (7.5 mmol/l).
A sustained haemoglobin level of greater than 12 g/dl should be avoided; guidance for appropriate
dose adjustment for when haemoglobin values exceeding 12 g/dl (7.5 mmol/l) are observed are
described below. A rise in haemoglobin of greater than 2 g/dL (1.25 mmol/l) over a four week period
should be avoided. If it occurs, appropriate dose adjustment should be made as provided.
Patients should be monitored closely to ensure that the lowest approved dose of Retacrit is used to
provide adequate control of the symptoms of anemia.
In patients with chronic renal failure and clinically evident ischemic heart disease or congestive heart
failure, maintenance haemoglobin concentration should not exceed the upper limit of the target
haemoglobin concentration.
Adult patients on haemodialysis
Retacrit should be administered either subcutaneously or intravenously.
The treatment is divided into two stages:
1. Correction phase: 50 IU/kg 3 times per week. When a dose adjustment is necessary, this should
be done in steps of at least four weeks. At each step, the increase or reduction
in dose should be of 25 IU/kg 3 times per week.
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 10 and 12 g/dl (6.2-7.5 mmol/l). The recommended total
weekly dose is between 75 and 300 IU/kg.
The clinical data available suggest that those patients whose initial haemoglobin is very low (< 6 g/dl
or < 3.75 mmol/l) may require higher maintenance doses than those whose initial anaemia is less
severe (Hb > 8 g/dl or > 5 mmol/l).
Paediatric patients on haemodialysis
The treatment is divided into two stages:
1. Correction phase 50 IU/kg, 3 times per week by the intravenous route. When a dose adjustment
is necessary, this should be done in steps of 25 IU/kg, 3 times per week at
intervals of at least 4 weeks until the desired goal is achieved.
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 9.5 and 11 g/dl (5.9-6.8 mmol/l).
Generally, children and adolescents under 30 kg body weight require higher maintenance doses than
adults and children over 30 kg. The following maintenance doses were observed in clinical trials after
6 months of treatment.
17
Dose (IU/kg given 3x week)
Weight (kg)
Median
Usual maintenance dose
< 10
100
75-150
10-30
75
60-150
> 30
33
30-100
The clinical data available suggest that those patients whose initial haemoglobin is very low
(< 6.8 g/dl or < 4.25 mmol/l) may require higher maintenance doses than those whose initial
haemoglobin is higher > 6.8 g/dl or> 4.25 mmol/l).
Adult patients on peritoneal dialysis
Retacrit should be administered either subcutaneously or intravenously.
The treatment is divided into two stages:
1. Correction phase: Starting dose of 50 IU/kg 2 times per week.
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 10 and 12 g/dl (6.2-7.5 mmol/l). Maintenance dose between
25 and 50 IU/kg 2 times per week into 2 equal doses.
Adult patients with renal insufficiency not yet undergoing dialysis
Retacrit should be administered either subcutaneously or intravenously.
The treatment is divided into two stages:
1. Correction phase: Starting dose of 50 IU/kg 3 times per week, followed if necessary by a dose
increase with 25 IU/kg increments (3 times per week) until the desired goal is
achieved (this should be done in steps of at least four weeks).
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 10 and 12 g/dl (6.2-7.5 mmol/l). Maintenance dose between
17 and 33 IU/kg 3 times per week.
The maximum dose should not exceed 200 IU/kg 3 times per week.
Retacrit should be administered by the subcutaneous route to patients with anaemia (e.g. haemoglobin
concentration ≤ 10 g/dl (6.2 mmol/l). Anaemia symptoms and sequelae may vary with age, gender,
and overall burden of disease; a physician´s evaluation of the individual patient´s clinical course and
condition is necessary.
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management with consideration for the haemoglobin target range of 10 g/dl (6.2 mmol/l)
to 12 g/dl (7.5 mmol/l). A sustained haemoglobin level of greater than 12 g/dl (7.5 mmol/l) should be
avoided; guidance for appropriate dose adjustment for when haemoglobin values exceeding 12 g/dl
(7.5 mmol/l) are observed are described below.
Patients should be monitored closely to ensure that the lowest approved dose of Retacrit is used to
provide adequate control of the symptoms of anaemia.
Retacrit therapy should continue until one month after the end of chemotherapy.
The initial dose is 150 IU/kg given subcutaneously 3 times per week. Alternatively, Retacrit can be
administered at an initial dose of 450 IU/kg subcutaneously once weekly.
18
Treatment of patients with chemotherapy induced anaemia










If the haemoglobin has increased by at least 1 g/dl (0.62 mmol/l) or the reticulocyte count has
increased ≥ 40,000 cells/µl above baseline after 4 weeks of treatment, the dose should remain at
150 IU/kg 3 times per week or 450 IU/kg once weekly. If the haemoglobin increase is < 1 g/dl
(< 0.62 mmol/l) and the reticulocyte count has increased < 40,000 cells/µl above baseline, increase the
dose to 300 IU/kg 3 times per week. If after an additional 4 weeks of therapy at 300 IU/kg 3 times per
week, the haemoglobin has increased ≥ 1 g/dl (0.62 mmol/l) or the reticulocyte count has increased
≥ 40,000 cells/µl the dose should remain at 300 IU/kg 3 times per week. However, if the haemoglobin
has increased < 1 g/dl (< 0.62 mmol/l) and the reticulocyte count has increased < 40,000 cells/µl above
baseline, response is unlikely and treatment should be discontinued.
The recommended dosing regimen is described in the following diagram:
Once the therapeutic objective for an individual patient has been achieved, the dose should be reduced
by 25 to 50% in order to maintain haemoglobin at that level. Appropriate dose titration should be
considered.
Dose adjustment
At a rate of rise in haemoglobin of > 2 g/dl (> 1.25 mmol/l) per month the Retacrit dose should be
reduced by about 25-50%. If haemoglobin level exceeds 12 g/dl (7.5 mmol/l), discontinue therapy
until it falls to 12 g/dl (7.5 mmol/l) or lower and then reinstitute Retacrit therapy at a dose 25% below
the previous dose.
−
Adult surgery patients in an autologous predonation programme.
Retacrit should be given by the intravenous route.
19

At the time of donating blood, Retacrit should be administered after the completion of the blood
donation procedure.
Mildly anaemic patients (haematocrit of 33-39%) requiring predeposit of ≥ 4 units of blood should be
treated with Retacrit at a dose of 600 IU/kg body weight 2 times weekly for 3 weeks prior to surgery.
All patients being treated with Retacrit should receive adequate iron supplementation (e.g. 200 mg oral
elemental iron daily) throughout the course of treatment. Iron supplementation should be started as
soon as possible, even several weeks prior to initiating the autologous predeposit, in order to achieve
high iron stores prior to starting Retacrit therapy.
Method of administration
For instructions on handling of the medicinal product before administration, see section 6.6.
The dose should be administered over at least 1-5 minutes, depending on the total dose. In
haemodialysed patients, a bolus injection may be given during the dialysis session through a suitable
venous port in the dialysis line. Alternatively, the injection can be given at the end of the dialysis
session via the fistula needle tubing, followed by 10 ml of sodium chloride 9 mg/ml (0.9%) solution
for injection to rinse the tubing and ensure satisfactory injection of the medicinal product into the
circulation.
A slower injection is preferable in patients who react to the treatment with “flu-like” symptoms.
Retacrit should not be administered by intravenous infusion.
Retacrit must not be mixed with other medicinal products (see section 6.2).
Subcutaneous injection
A maximum volume of 1 ml at one injection site should generally not be exceeded. In case of larger
volumes, more than one site should be chosen for the injection.
The injections are given in the limbs or the anterior abdominal wall.
4.3
−
Hypersensitivity to the active substance or to any of the excipients.
−
Patients who develop Pure Red Cell Aplasia (PRCA) following treatment with any
erythropoietin must not receive Retacrit or any other erythropoietin (see section 4.4).
−
Uncontrolled hypertension.
−
In the indication "increasing the yield of autologous blood": myocardial infarction or stroke in
the month preceding treatment, unstable angina pectoris, increased risk of deep venous
thrombosis such as history of venous thromboembolic disease.
−
Patients who for any reason cannot receive adequate antithrombotic prophylaxis.
4.4
General
Like in all patients receiving erythropoietin, blood pressure may rise during treatment with Retacrit.
Blood pressure should be closely monitored and adequately controlled in all epoetin treatment naïve as
well as pre-treated patients before, at initiation of, and during treatment with Retacrit. It may be
necessary to add or increase anti-hypertensive treatment. If blood pressure cannot be well controlled,
Retacrit treatment should be discontinued.
Retacrit should also be used with caution in the presence of epilepsy and chronic liver failure.
20
Intravenous injection
There may be a moderate dose-dependent rise in the platelet count within the normal range during
treatment with erythropoietin. This regresses during the course of continued therapy. It is
recommended that the platelet count is regularly monitored during the first 8 weeks of therapy.
All other causes of anaemia (iron deficiency, haemolysis, blood loss, vitamin B
12
- or folate
deficiencies) should be considered and treated prior to initiating and during therapy with Retacrit. In
most cases, the ferritin values in the serum fall simultaneously with the rise in packed cell volume. In
order to ensure optimum response to erythropoietin, adequate iron stores should be assured:
−
iron supplementation, e.g. 200-300 mg/day orally (100-200 mg/day for paediatric patients) is
recommended for chronic renal failure patients whose serum ferritin levels are below 100 ng/ml
−
oral iron substitution of 200-300 mg/day is recommended for all cancer patients whose
transferrin saturation is below 20%.
All of these additive factors of anaemia should also be carefully considered when deciding to increase
the dose of erythropoietin in cancer patients.
A paradoxical decrease in haemoglobin and development of severe anaemia associated with low
reticulocyte counts should prompt to discontinue treatment with epoetin and perform anti-
erythropoietin antibody testing. Cases have been reported in patients with hepatits C treated with
interferon and ribavirin, when epoetins are used concomitantly. Epoetins are not approved in the
management of anaemia associated with hepatitis C.
In order to improve the traceability of ESAs, the name of the prescribed ESA should be clearly
recorded (or: stated) in the patient file.
Good blood management practices should always be used in the perisurgical setting.
This medicinal product contains phenylalanine which may be harmful for people with
phenylketonuria.
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, i.e. essentially
‘sodium-free’.
Chronic renal failure patients
Haemoglobin concentration
In patients with chronic renal failure, maintenance haemoglobin concentration should not exceed the
upper limit of the target haemoglobin concentration recommended in section 4.2. In clinical trials, an
increased risk of death, serious cardiovascular events or cerebrovascular events including stroke were
observed when ESAs were administered to target a haemoglobin of greater than 12 g/dl (7.5 mmol/l).
Controlled clinical trials have not shown significant benefits attributable to the administration of
epoetins when haemoglobin concentration is increased beyond the level necessary to control
symptoms of anaemia and to avoid blood transfusion.
Haemoglobin levels should be measured on a regular basis until a stable level is achieved and
periodically thereafter. The rate of increase in haemoglobin should be approximately 1 g/dl
(0.62 mmol/l) per month and should not exceed 2 g/dl (1.25 mmol/l) per month to minimize the risk of
developing or worsening of hypertension.
Chronic renal failure patients treated with Retacrit by the subcutaneous route should be monitored
regularly for loss of efficacy, defined as absent or decreased response to Retacrit treatment in patients
who previously responded to such therapy. This is characterised by a sustained decrease in
haemoglobin despite an increase in Retacrit dosage.
21
Non response to erythropoietin therapy should prompt a search for causative factors. These include:
iron, folate, or Vitamin B
12
deficiency; aluminium intoxication; intercurrent infections; inflammatory
or traumatic episodes; occult blood loss; haemolysis, and bone marrow fibrosis of any origin.
Cases of antibody-mediated PRCA have been very rarely reported in chronic renal failure patients
with erythropoietin administered by the subcutaneous route. In patients developing sudden lack of
efficacy, defined by a decrease in haemoglobin (1-2 g/dl per month) with increased need for
transfusions, a reticulocyte count should be obtained and typical causes of non-response (e.g. iron,
folate, or Vitamin B
12
-deficiency, aluminium intoxication, infection or inflammation, blood loss, and
haemolysis) should be investigated. If no cause is identified, a bone marrow examination should be
considered for diagnosis of PRCA.
If PRCA is diagnosed, therapy with Retacrit must be immediately discontinued and testing for
erythropoietin antibodies should be considered. Patients should not be switched to another medicinal
product as anti-erythropoietin antibodies cross-react with other erythropoietins. Other causes of PRCA
should be excluded, and appropriate therapy initiated.
Monitoring of reticulocyte count on a regular basis is recommended to detect possible occurrence of
lack of efficacy in chronic renal failure patients.
Hyperkalaemia has been observed in isolated cases. In chronic renal failure patients, correction for
anaemia may lead to increased appetite, and potassium and protein intake. Dialysis prescriptions may
have to be adjusted periodically to maintain urea, creatinine and potassium in the desired range. Serum
electrolytes should be monitored in chronic renal failure patients. If an elevated (or rising) serum
potassium level is detected then consideration should be given to ceasing erythropoietin administration
until hyperkalaemia has been corrected.
An increase in heparin dose during haemodialysis is frequently required during the course of therapy
with erythropoietin as a result of the increased packed cell volume. Occlusion of the dialysis system is
possible if heparinisation is not optimum.
Based on information available to date, correction of anaemia with erythropoietin in adult patients
with renal insufficiency not yet undergoing dialysis does not accelerate the rate of progression of renal
insufficiency.
Adult cancer patients with symptomatic anaemia receiving chemotherapy
In cancer patients receiving chemotherapy, the 2-3 week delay between erythropoietin administration
and the appearance of erythropoietin-induced red cells should be taken into account when assessing if
Retacrit therapy is appropriate (patient at risk of being transfused).
Haemoglobin levels should be closely monitored until a stable level is achieved and periodically
thereafter. If the rate of increase in haemoglobin exceeds 2 g/dl (1.25 mmol/l) per month or the
haemoglobin level exceeds 12 g/dl (7,5 mmol/l), the dose adjustment detailed in section 4.2 should be
thoroughly performed to minimise the risk of thrombotic events (see section 4.2).
As an increased incidence of thrombotic vascular events (TVEs) has been observed in cancer patients
receiving erythropoietic agents (see section 4.8), this risk should be carefully weighed against the
benefit to be derived from treatment (with Retacrit) particularly in cancer patients with an increased
risk of thrombotic vascular events, such as obesity and patients with a prior history of TVEs (e.g. deep
venous thrombosis or pulmonary embolism).
Adult surgery patients in an autologous predonation programme
All special warnings and precautions associated with autologous predonation programs, especially
routine volume replacement, should be respected.
22
Tumour growth potential
Epoetins are growth factors that primarily stimulate red blood cell production. Erythropoietin
receptors may be expressed on the surface of a variety of tumour cells. As with all growth factors,
there is a concern that epoetins could stimulate the growth of any type of malignancy. In several
controlled studies, epoetins have not been shown to improve overall survival or decrease the risk of
tumour progression in patients with anaemia associated with cancer.
Several controlled clinical studies in which epoetins were administered to patients with a variety of
common tumours including squamous head and neck cancer, lung cancer, and breast cancer, have
shown an unexplained excess mortality.
In controlled clinical studies, use of Epoetin alfa and other erythropoiesis-stimulating agents (ESAs)
have shown:
•
shortened time to tumour progression in patients with advanced head and neck cancer
receiving radiation therapy when administered to target a haemoglobin of greater than 14 g/dl
(8.7 mmol/l),
•
shortened overall survival and increased deaths attributed to disease progression at 4 months
in patients with metastatic breast cancer receiving chemotherapy when administered to target a
haemoglobin of 12-14 g/dl (7.5 -8.7 mmol/l),
•
increased risk of death when administered to target a haemoglobin of 12 g/dl (7.5 mmol/l) in
patients with active malignant disease receiving neither chemotherapy nor radiation therapy.
ESAs are not indicated for use in this patient population.
In view of the above, in some clinical situations blood transfusion should be the preferred treatment
for the management of anaemia in patients with cancer. The decision to administer recombinant
erythropoietins should be based on a benefit-risk assessment with the participation of the individual
patient, which should take into account the specific clinical context. Factors that should be considered
in this assessment should include the type of tumour and its stage; the degree of anaemia; life-
expectancy; the environment in which the patient is being treated; and patient preference (see section
5.1).
4.5
There is no evidence to indicate that treatment with erythropoietin alters the metabolism of other
medicinal products.
However, since ciclosporin is bound by red blood cells there is potential for interactions with other
medicinal products. If erythropoietin is given concomitantly with ciclosporin, blood levels of
ciclosporin should be monitored and the dose of ciclosporin adjusted as the haematocrit rises.
No evidence exists that indicates an interaction between epoetin alfa and G-CSF or GM-CSF with
regard to haematological differentiation or proliferation of tumour biopsy specimens in vitro.
4.6
There are no adequate and well-controlled studies in pregnant women. Studies in animals have shown
reproduction toxicity (see section 5.3). Consequently, erythropoietin should generally be used during
pregnancy and lactation only if the potential benefit outweighs the potential risk to the foetus.
4.7
Retacrit has no or negligible influence on the ability to drive and use machines.
4.8
23
Retacrit is a biological medicinal product. Data from clinical studies with Retacrit are in line with the
safety profile of other authorized erythropoietins. Based on the results from clinical trials with other
authorized erythropoietins approximately 8% of patients treated with erythropoietin are expected to
predominantly in patients with chronic renal failure or underlying malignancies. These undesirable
effects are most commonly headache and a dose dependent increase in blood pressure. Hypertensive
crisis with encephalopathy-like symptoms can occur. Attention should be paid to sudden stabbing
migraine-like headaches as a possible warning signal.
Thrombotic/vascular events, such as myocardial ischaemia, myocardial infarction, cerebrovascular
accidents (cerebral haemorrhage and cerebral infarction), transient ischaemic attacks, deep vein
thrombosis, arterial thrombosis, pulmonary emboli, aneurysms, retinal thrombosis, and clotting of an
artificial kidney have been reported in patients receiving erythropoietic agents.
Antibody-mediated erythroblastopenia (PRCA) has been reported after months to years of treatment
with epoetin alfa. In most of these patients, antibodies to erythropoietins have been observed (see
sections 4.3 and 4.4).
In this section frequencies of undesirable effects are defined as follows: Very common (
>
1/10);
common (
>
1/100 to <1/10); uncommon (
>
1/1,000 to <1/100); rare (
>
1/10,000 to <1/1,000); very rare
(<1/10,000), not known (frequency cannot be estimated from the available data).
SOC
Frequency
ADR
very rare
Thrombocytosis (see section 4.4)
Blood and lymphatic system
disorders
Frequency not known
Antibody-mediated erythroblastopenia
(PRCA)
Immune system disorders
rare
Hypersensitivity reactions
very rare
anaphylactic reaction
very common
dizziness (chronic renal failure
patients)
headache (cancer patients)
Nervous system disorders
common
dizziness (cancer patients)
headache (chronic renal failure
patients)
stroke
uncommon
cerebral haemorrhage
Frequency not known
cerebral infarction
transient ischaemic attacks
hypertensive encephalopathy
Eye disorders
Frequency not known
retinal thrombosis
Cardiac disorders
Frequency not known
myocardial infarction
myocardial ischaemia
common
deep vein thrombosis (cancer patients)
increase in blood pressure
Vascular disorders
aneurysms
arterial thrombosis
deep vein thrombosis (chronic renal
failure patients)
hypertensive crisis
Frequency not known
Respiratory, thoracic and
mediastinal disorders
common
pulmonary embolism (cancer patients)
Frequency not known
pulmonary embolism (chronic renal
failure patients)
Skin and subcutaneous tissue
disorders
common
Non-specific skin rashes
very rare
Angioedema
24












































Frequency not known
pruritus
very common
joint pains (chronic renal failure
patients)
Musculoskeletal and
connective tissue disorders
common
joint pains (cancer patients)
very common
"Flu-like" symptoms (chronic renal
failure patients)
feelings of weakness (chronic renal
failure patients)
tiredness (chronic renal failure
patients)
General disorders and
administration site conditions
common
"Flu-like" symptoms (cancer patients)
feelings of weakness (cancer patients)
tiredness (cancer patients)
Injury, poisoning and
procedural complications
common
clotting of an artificial kidney
Adult and paediatric haemodialysis patients, adult peritoneal dialysis patients and adult patients with
renal insufficiency not yet undergoing dialysis
The most frequent adverse reaction during treatment with epoetin alfa is a dose-dependent increase in
blood pressure or aggravation of existing hypertension. These increases in blood pressure can be
treated with medicinal products. Moreover, monitoring of the blood pressure is recommended
particularly at the start of therapy. The following reactions have also occurred in isolated patients with
normal or low blood pressure: hypertensive crisis with encephalopathy-like symptoms (e.g. headaches
and confused state) and generalised tonoclonal seizures, requiring the immediate attention of a
physician and intensive medical care. Particular attention should be paid to sudden stabbing migraine
like headaches as a possible warning signal.
Shunt thromboses may occur, especially in patients who have a tendency to hypotension or whose
arteriovenous fistulae exhibit complications (e.g. stenoses, aneurysms, etc.). Early shunt revision and
thrombosis prophylaxis by administration of acetylsalicylic acid, for example, is recommended in
these patients.
Adult cancer patients with symptomatic anaemia receiving chemotherapy
Hypertension may occur in epoetin alfa treated patients. Consequently, haemoglobin and blood
pressure should be closely monitored.
An increased incidence of thrombotic vascular events (see section 4.4 and section 4.8 - General) has
been observed in patients receiving erythropoietic agents.
Surgery patients in autologous predonation programmes
Independent of erythropoietin treatment, thrombotic and vascular events may occur in surgical patients
with underlying cardiovascular disease following repeated phlebotomy. Therefore, routine volume
replacement should be performed in such patients.
4.9
The therapeutic margin of erythropoietin is very wide. Overdose of erythropoietin may produce effects
that are extensions of the pharmacological effects of the hormone. Phlebotomy may be performed if
excessively high haemoglobin levels occur. Additional supportive care should be provided as
necessary.
25




















5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Antianaemic preparations, erythropoietin
ATC code: B03XA01
Retacrit is a biosimilar medicinal product. Detailed information is available on the website of the
European Medicines Agency http://www.ema.europa.eu.
Erythropoietin is a glycoprotein that stimulates, as a mitosis-stimulating factor and differentiating
hormone, the formation of erythrocytes from precursors of the stem cell compartment.
The apparent molecular weight of erythropoietin is 32,000-40,000 Dalton. The protein moiety of the
molecule contributes about 58% of total molecular weight and consists of 165 amino acids. The four
carbohydrate chains are attached via three N-glycosidic bonds and one O-glycosidic bond to the
protein. Epoetin zeta is identical in its amino acid sequence and similar in carbohydrate composition to
endogenous human erythropoietin that has been isolated from the urine of anaemic patients.
The biological efficacy of erythropoietin has been demonstrated in various animal models
in vivo
(normal and anaemic rats, polycythaemic mice). After administration of erythropoietin, the number of
erythrocytes, the Hb values and reticulocyte counts increase as well as the
59
Fe-incorporation rate.
An increased
3
H-thymidine incorporation in the erythroid nucleated spleen cells has been found
in vitro
(mouse spleen cell culture) after incubation with erythropoietin. It could be shown with the aid
of cell cultures of human bone marrow cells that erythropoietin stimulates erythropoiesis specifically
and does not affect leucopoiesis. Cytotoxic actions of erythropoietin on bone marrow cells could not
be detected.
As with other haematopoietic growth factors, erythropoietin has shown
in vitro
stimulating properties
on human endothelial cells.
721 cancer patients receiving non-platinum chemotherapy were included in three placebo-controlled
studies, 389 patients with haematological malignancies (221 multiple myeloma, 144 non-Hodgkin's
lymphoma, and 24 other haematological malignancies) and 332 with solid tumours (172 breast,
64 gynaecological, 23 lung, 22 prostate, 21 gastrointestinal, and 30 other tumour types). In two large,
open-label studies, 2697 cancer patients receiving non-platinum chemotherapy were included,
1895 with solid tumours (683 breast, 260 lung, 174 gynaecological, 300 gastrointestinal, and 478 other
tumour types) and 802 with haematological malignancies.
In a prospective, randomised, double-blind, placebo-controlled trial conducted in 375 anaemic patients
with various non-myeloid malignancies receiving non-platinum chemotherapy, there was a significant
reduction of anaemia-related sequelae (e.g. fatigue, decreased energy, and activity reduction), as
measured by the following instruments and scales: Functional Assessment of Cancer
Therapy-Anaemia (FACT-An) general scale, FACT-An fatigue scale, and Cancer Linear Analogue
Scale (CLAS). Two other smaller, randomized, placebo-controlled trials failed to show a significant
improvement in quality of life parameters on the EORTC-QLQ-C30 scale or CLAS, respectively.
Erythropoietin is a growth factor that primarily stimulates red cell production. Erythropoietin receptors
may be expressed on the surface of a variety of tumour cells.
Survival and tumour progression have been examined in five large controlled studies involving a total
of 2833 patients, of which four were double-blind placebo-controlled studies and one was an open-
label study. The studies either recruited patients who were being treated with chemotherapy (two
studies) or used patient populations in which erythropoiesis stimulating agents are not indicated:
anaemia in patients with cancer not receiving chemotherapy, and head and neck cancer patients
26
receiving radiotherapy. The target haemoglobin concentration in two studies was > 13 g/dl; in the
remaining three studies it was 12-14 g/dl. In the open-label study there was no difference in overall
survival between patients treated with recombinant human erythropoietin and controls. In the four
placebo-controlled studies the hazard ratios for overall survival ranged between 1.25 and 2.47 in
favour of controls. These studies have shown a consistent unexplained statistically significant excess
mortality in patients who have anaemia associated with various common cancers who received
recombinant human erythropoietin compared to controls. Overall survival outcome in the trials could
not be statisfactorily explained by differences in the incidence of thrombosis and related complications
between those given recombinant human erythropoietin and those in the control group.
A systematic review has also been performed involving more than 9000 cancer patients participating
in 57 clinical trials. Meta-analysis of overall survival data produced a hazard ratio point estimate of
1.08 in favour of controls (95% CI: 0.99, 1,18; 42 trials and 8167 patients). An increased relative risk
of thromboembolic events (RR 1.67, 95% CI: 1.35, 2.06, 35 trials and 6769 patients) was observed in
patients treated with recombinant human erythropoietin. There is an increased risk for
thromboembolic events in patients with cancer treated with recombinant human erythropoietin and a
negative impact on overall survival cannot be excluded. The extent to which these outcomes might
apply to the administration of recombinant human erythropoietin to patients with cancer, treated with
chemotherapy to achieve haemoglobin concentrations less than 13 g/dl, is unclear because few patients
with these characteristics were included in the data reviewed.
A patient-level data analysis has also been performed on more than 13.900 cancer patients (chemo-,
radio-, chemoradio-, or no therapy) participating in 53 controlled clinical trials involving several
epoetins. Meta-analysis of overall survival data produced a hazard ratio point estimate of 1.06 in
favour of controls (95% CI: 1.00, 1.12; 53 trials and 13933 patients) and for the cancer patients
receiving chemotherapy, the overall survival hazard ratio was 1.04 (95% CI: 0.97, 1.11; 38 trials and
10.441 patients). Meta-analyses also indicate consistently a significantly increased relative risk of
thromboembolic events in cancer patients receiving recombinant human erythropoietin (see section
4.4).
In a randomised, double-blind, placebo-controlled study of 4,038 CRF patients not on dialysis with
type 2 diabetes and haemoglobin levels ≤ 11 g/dL, patients received either treatment with darbepoetin
alfa to target haemoglobin levels of 13 g/dL or placebo (see section 4.4). The study did not meet either
primary objective of demonstrating a reduction in risk for all-cause mortality, cardiovascular
morbidity, or end stage renal disease (ESRD). Analysis of the individual components of the composite
endpoints showed the following HR (95% CI): death 1.05 (0.92, 1.21), stroke 1.92 (1.38, 2.68),
congestive heart failure (CHF) 0.89 (0.74, 1.08), myocardial infarction (MI) 0.96 (0.75, 1.23),
hospitalisation for myocardial ischaemia 0.84 (0.55, 1.27), ESRD 1.02 (0.87, 1.18).
5.2
Pharmacokinetic properties
Measurement of erythropoietin following multiple dose intravenous administration revealed a half-life
of approximately 4 hours in healthy volunteers and a somewhat more prolonged half-life of
approximately 5 hours in renal failure patients. A half-life of approximately 6 hours has been reported
in children.
Subcutaneous route
Following subcutaneous injection, serum levels of erythropoietin are much lower than the levels
achieved following intravenous injection, the levels increase slowly and reach a peak between 12 and
18 hours postdose. The peak is always well below the peak achieved using the intravenous route
(approximately 1/20th of the value).
There is no accumulation: the levels remain the same, whether they are determined 24 hours after the
first injection or 24 hours after the last injection.
27
Intravenous route
The half-life is difficult to evaluate for the subcutaneous route and is estimated to be about 24 hours.
The bioavailability of subcutaneous injectable erythropoietin is much lower than that of the
intravenous medicinal product and is approximately 20%.
5.3
Preclinical safety data
In some pre-clinical toxicological studies in dogs and rats, but not in monkeys, erythropoietin therapy
was associated with subclinical bone marrow fibrosis (bone marrow fibrosis is a known complication
of chronic renal failure in humans and may be related to secondary hyperparathyroidism or unknown
factors. The incidence of bone marrow fibrosis was not increased in a study of haemodialysis patients
who were treated with erythropoietin for 3 years compared to a matched control group of dialysis
patients who had not been treated with erythropoietin).
In animal studies, erythropoietin has been shown to decrease foetal body weight, delay ossification
and increase foetal mortality when given in weekly doses of approximately 20 times the recommended
human weekly dose. These changes are interpreted as being secondary to decreased maternal body
weight gain.
Erythropoietin did not show any changes in bacterial and mammalian cell culture mutagenicity tests
and an
in vivo
micronucleus test in mice. Long-term carcinogenicity studies have not been carried out.
There are conflicting reports in the literature regarding whether erythropoietin may play a major role
as tumour proliferator. These reports are based on
in vitro
findings from human tumour samples, but
are of uncertain significance in the clinical situation.
6.
6.1
List of Excipients
Disodium phosphate dihydrate
Sodium dihydrogen phosphate dihydrate
Sodium chloride
Calcium chloride dihydrate
Polysorbate 20
Glycine
Leucine
Isoleucine
Threonine
Glutamic acid
Phenylalanine
Water for injections
Sodium hydroxide (pH adjuster)
Hydrochloric acid (pH adjuster)
6.2
Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3
Shelf life
2 years
6.4
Special precautions for storage
Store in a refrigerator (2°C - 8°C). Do not freeze.
28
Keep the pre-filled syringe in the outer carton in order to protect from light.
For the purpose of ambulatory use, the patient may remove the product from the refrigerator and store
it at room temperature (not above 25°C) for one single period of up to 3 days.
6.5
Nature and contents of container
Pre-filled syringe Type I glass with a fixed steel injection needle and a plunger stopper with PTFE
coating.
Each pre-filled syringe contains 0.6 ml solution for injection.
Each pack contains 1 or 6 pre-filled syringes.
Not all pack sizes may be marketed.
6.6
Special precautions for disposal and other handling
Handling instructions for Retacrit:
1.
After removing one syringe from the blister pack the solution should be checked to ensure that
it is clear, colourless and practically free from visible particles.
2.
The protective cap is removed from the injection needle and air is expelled from the syringe and
needle by holding the syringe vertically and gently pressing the plunger upwards.
3.
The syringe is now ready for use.
Retacrit must not be used if
•
The blister sealing is broken or the blister is damaged in any way.
•
The liquid is coloured or you can see particles floating in it.
•
Any liquid has leaked out of the pre-filled syringe or condensation is visible within the sealed
blister.
•
It may have been accidentally frozen.
This medicinal product is for single use only.
Do not shake.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Hospira UK Limited
Queensway
Royal Leamington Spa
Warwickshire
CV31 3RW
United Kingdom
8.
EU/1/07/431/003
EU/1/07/431/004
9.
29
1.
Retacrit 3000 IU/0.9ml solution for injection in pre-filled syringe
2.
1 pre-filled syringe with 0.9 ml solution for injection contains 3000 international units (IU) epoetin
zeta* (recombinant human erythropoietin). The solution contains 3333 IU Epoetin zeta per ml.
*Produced by recombinant DNA technology in Chinese Hamster Ovary (CHO) cell line.
Excipient:
Each pre-filled syringe contains 0.45 mg phenylalanine.
For a full list of excipients, see section 6.1.
3.
Solution for injection in pre-filled syringe.
Clear, colourless solution.
4.
4.1
−
Treatment of symptomatic anaemia associated with chronic renal failure (CRF) in adult and
paediatric patients:
o
Treatment of anaemia associated with chronic renal failure in adult and paediatric
patients on haemodialysis and adult patients on peritoneal dialysis (See section 4.4).
o
Treatment of severe anaemia of renal origin accompanied by clinical symptoms in
adult patients with renal insufficiency not yet undergoing dialysis (See section 4.4).
−
Treatment of anaemia and reduction of transfusion requirements in adult patients receiving
chemotherapy for solid tumours, malignant lymphoma or multiple myeloma, and at risk of
transfusion as assessed by the patient's general status (e.g. cardiovascular status, pre-existing
anaemia at the start of chemotherapy).
−
Retacrit can be used to increase the yield of autologous blood from patients in a predonation
programme. Its use in this indication must be balanced against the reported risk of
thromboembolic events. Treatment should only be given to patients with moderate anaemia (no
iron deficiency), if blood saving procedures are not available or insufficient when the scheduled
major elective surgery requires a large volume of blood (4 or more units of blood for females or
5 or more units for males).
4.2
Treatment with Retacrit has to be initiated under the supervision of physicians experienced in the
management of patients with above indications.
Posology
Treatment of symptomatic anaemia in adult and paediatric chronic renal failure patients
Retacrit should be administered either subcutaneously or intravenously.
31
The haemoglobin concentration aimed for is between 10 and 12 g/dl (6.2-7.5 mmol/l), except in
paediatric patients in whom the haemoglobin concentration should be between 9.5 and 11 g/dl
(5.9-6.8 mmol/l). The upper limit of the target haemoglobin concentration should not be exceeded.
Anemia symptoms and sequaelea may vary with age, gender and overall burden of disesase; a
physician´s evaluation of the individual patient´s clinical course and condition is necessary. Retacrit
should be administered either subcutaneously or intravenously in order to increase haemoglobin to not
greater than 12 g/dL (7.5 mmol/L) Due to intra-patient variability, occasional individual haemoglobin
values for a patient above and below the desired haemoglobin level may be observed. Haemoglobin
variability should be addressed through dose management, with consideration for the haemoglobin
target range of 10 g/dL (6.2 mmol/l) to 12 g/dl (7.5 mmol/l).
A sustained haemoglobin level of greater than 12 g/dl should be avoided; guidance for appropriate
dose adjustment for when haemoglobin values exceeding 12 g/dl (7.5 mmol/l) are observed are
described below. A rise in haemoglobin of greater than 2 g/dL (1.25 mmol/l) over a four week period
should be avoided. If it occurs, appropriate dose adjustment should be made as provided.
Patients should be monitored closely to ensure that the lowest approved dose of Retacrit is used to
provide adequate control of the symptoms of anemia.
In patients with chronic renal failure and clinically evident ischemic heart disease or congestive heart
failure, maintenance haemoglobin concentration should not exceed the upper limit of the target
haemoglobin concentration.
Adult patients on haemodialysis
Retacrit should be administered either subcutaneously or intravenously.
The treatment is divided into two stages:
1. Correction phase: 50 IU/kg 3 times per week. When a dose adjustment is necessary, this should
be done in steps of at least four weeks. At each step, the increase or reduction
in dose should be of 25 IU/kg 3 times per week.
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 10 and 12 g/dl (6.2-7.5 mmol/l). The recommended total
weekly dose is between 75 and 300 IU/kg.
The clinical data available suggest that those patients whose initial haemoglobin is very low (< 6 g/dl
or < 3.75 mmol/l) may require higher maintenance doses than those whose initial anaemia is less
severe (Hb > 8 g/dl or > 5 mmol/l).
Paediatric patients on haemodialysis
The treatment is divided into two stages:
1. Correction phase 50 IU/kg, 3 times per week by the intravenous route. When a dose adjustment
is necessary, this should be done in steps of 25 IU/kg, 3 times per week at
intervals of at least 4 weeks until the desired goal is achieved.
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 9.5 and 11 g/dl (5.9-6.8 mmol/l).
Generally, children and adolescents under 30 kg body weight require higher maintenance doses than
adults and children over 30 kg. The following maintenance doses were observed in clinical trials after
6 months of treatment.
32
Dose (IU/kg given 3x week)
Weight (kg)
Median
Usual maintenance dose
< 10
100
75-150
10-30
75
60-150
> 30
33
30-100
The clinical data available suggest that those patients whose initial haemoglobin is very low
(< 6.8 g/dl or < 4.25 mmol/l) may require higher maintenance doses than those whose initial
haemoglobin is higher > 6.8 g/dl or> 4.25 mmol/l).
Adult patients on peritoneal dialysis
Retacrit should be administered either subcutaneously or intravenously.
The treatment is divided into two stages:
1. Correction phase: Starting dose of 50 IU/kg 2 times per week.
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 10 and 12 g/dl (6.2-7.5 mmol/l). Maintenance dose between
25 and 50 IU/kg 2 times per week into 2 equal doses.
Adult patients with renal insufficiency not yet undergoing dialysis
Retacrit should be administered either subcutaneously or intravenously.
The treatment is divided into two stages:
1. Correction phase: Starting dose of 50 IU/kg 3 times per week, followed if necessary by a dose
increase with 25 IU/kg increments (3 times per week) until the desired goal is
achieved (this should be done in steps of at least four weeks).
2. Maintenance phase: Dose adjustment in order to maintain haemoglobin (Hb) values at the desired
level: Hb between 10 and 12 g/dl (6.2-7.5 mmol/l). Maintenance dose between
17 and 33 IU/kg 3 times per week.
The maximum dose should not exceed 200 IU/kg 3 times per week.
Treatment of patients with chemotherapy induced anaemia
Retacrit should be administered by the subcutaneous route to patients with anaemia (e.g. haemoglobin
concentration ≤ 10 g/dl (6.2 mmol/l). Anaemia symptoms and sequelae may vary with age, gender,
and overall burden of disease; a physician´s evaluation of the individual patient´s clinical course and
condition is necessary.
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and
below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed
through dose management with consideration for the haemoglobin target range of 10 g/dl (6.2 mmol/l)
to 12 g/dl (7.5 mmol/l). A sustained haemoglobin level of greater than 12 g/dl (7.5 mmol/l) should be
avoided; guidance for appropriate dose adjustment for when haemoglobin values exceeding 12 g/dl
(7.5 mmol/l) are observed are described below.
Patients should be monitored closely to ensure that the lowest approved dose of Retacrit is used to
provide adequate control of the symptoms of anaemia.
Retacrit therapy should continue until one month after the end of chemotherapy.
The initial dose is 150 IU/kg given subcutaneously 3 times per week. Alternatively, Retacrit can be
administered at an initial dose of 450 IU/kg subcutaneously once weekly.
33










If the haemoglobin has increased by at least 1 g/dl (0.62 mmol/l) or the reticulocyte count has
increased ≥ 40,000 cells/µl above baseline after 4 weeks of treatment, the dose should remain at
150 IU/kg 3 times per week or 450 IU/kg once weekly. If the haemoglobin increase is < 1 g/dl
(< 0.62 mmol/l) and the reticulocyte count has increased < 40,000 cells/µl above baseline, increase the
dose to 300 IU/kg 3 times per week. If after an additional 4 weeks of therapy at 300 IU/kg 3 times per
week, the haemoglobin has increased ≥ 1 g/dl (0.62 mmol/l) or the reticulocyte count has increased
≥ 40,000 cells/µl the dose should remain at 300 IU/kg 3 times per week. However, if the haemoglobin
has increased < 1 g/dl (< 0.62 mmol/l) and the reticulocyte count has increased < 40,000 cells/µl above
baseline, response is unlikely and treatment should be discontinued.
The recommended dosing regimen is described in the following diagram:
Once the therapeutic objective for an individual patient has been achieved, the dose should be reduced
by 25 to 50% in order to maintain haemoglobin at that level. Appropriate dose titration should be
considered.
Dose adjustment
At a rate of rise in haemoglobin of > 2 g/dl (> 1.25 mmol/l) per month the Retacrit dose should be
reduced by about 25-50%. If haemoglobin level exceeds 12 g/dl (7.5 mmol/l), discontinue therapy
until it falls to 12 g/dl (7.5 mmol/l) or lower and then reinstitute Retacrit therapy at a dose 25% below
the previous dose.
−
Adult surgery patients in an autologous predonation programme.
Retacrit should be given by the intravenous route.
34

At the time of donating blood, Retacrit should be administered after the completion of the blood
donation procedure.
Mildly anaemic patients (haematocrit of 33-39%) requiring predeposit of ≥ 4 units of blood should be
treated with Retacrit at a dose of 600 IU/kg body weight 2 times weekly for 3 weeks prior to surgery.
All patients being treated with Retacrit should receive adequate iron supplementation (e.g. 200 mg oral
elemental iron daily) throughout the course of treatment. Iron supplementation should be started as
soon as possible, even several weeks prior to initiating the autologous predeposit, in order to achieve
high iron stores prior to starting Retacrit therapy.
Method of administration
For instructions on handling of the medicinal product before administration, see section 6.6.
The dose should be administered over at least 1-5 minutes, depending on the total dose. In
haemodialysed patients, a bolus injection may be given during the dialysis session through a suitable
venous port in the dialysis line. Alternatively, the injection can be given at the end of the dialysis
session via the fistula needle tubing, followed by 10 ml of sodium chloride 9 mg/ml (0.9%) solution
for injection to rinse the tubing and ensure satisfactory injection of the medicinal product into the
circulation.
A slower injection is preferable in patients who react to the treatment with “flu-like” symptoms.
Retacrit should not be administered by intravenous infusion.
Retacrit must not be mixed with other medicinal products (see section 6.2).
Subcutaneous injection
A maximum volume of 1 ml at one injection site should generally not be exceeded. In case of larger
volumes, more than one site should be chosen for the injection.
The injections are given in the limbs or the anterior abdominal wall.
4.3
−
Hypersensitivity to the active substance or to any of the excipients.
−
Patients who develop Pure Red Cell Aplasia (PRCA) following treatment with any
erythropoietin must not receive Retacrit or any other erythropoietin (see section 4.4).
−
Uncontrolled hypertension.
−
In the indication "increasing the yield of autologous blood": myocardial infarction or stroke in
the month preceding treatment, unstable angina pectoris, increased risk of deep venous
thrombosis such as history of venous thromboembolic disease.
−
Patients who for any reason cannot receive adequate antithrombotic prophylaxis.
4.4
General
Like in all patients receiving erythropoietin, blood pressure may rise during treatment with Retacrit.
Blood pressure should be closely monitored and adequately controlled in all epoetin treatment naïve as
well as pre-treated patients before, at initiation of, and during treatment with Retacrit. It may be
necessary to add or increase anti-hypertensive treatment. If blood pressure cannot be well controlled,
Retacrit treatment should be discontinued.
Retacrit should also be used with caution in the presence of epilepsy and chronic liver failure.
35
Intravenous injection
There may be a moderate dose-dependent rise in the platelet count within the normal range during
treatment with erythropoietin. This regresses during the course of continued therapy. It is
recommended that the platelet count is regularly monitored during the first 8 weeks of therapy.
All other causes of anaemia (iron deficiency, haemolysis, blood loss, vitamin B
12
- or folate
deficiencies) should be considered and treated prior to initiating and during therapy with Retacrit. In
most cases, the ferritin values in the serum fall simultaneously with the rise in packed cell volume. In
order to ensure optimum response to erythropoietin, adequate iron stores should be assured:
−
iron supplementation, e.g. 200-300 mg/day orally (100-200 mg/day for paediatric patients) is
recommended for chronic renal failure patients whose serum ferritin levels are below 100 ng/ml
−
oral iron substitution of 200-300 mg/day is recommended for all cancer patients whose
transferrin saturation is below 20%.
All of these additive factors of anaemia should also be carefully considered when deciding to increase
the dose of erythropoietin in cancer patients.
A paradoxical decrease in haemoglobin and development of severe anaemia associated with low
reticulocyte counts should prompt to discontinue treatment with epoetin and perform anti-
erythropoietin antibody testing. Cases have been reported in patients with hepatits C treated with
interferon and ribavirin, when epoetins are used concomitantly. Epoetins are not approved in the
management of anaemia associated with hepatitis C.
In order to improve the traceability of ESAs, the name of the prescribed ESA should be clearly
recorded (or: stated) in the patient file.
Good blood management practices should always be used in the perisurgical setting.
This medicinal product contains phenylalanine which may be harmful for people with
phenylketonuria.
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, i.e. essentially
‘sodium-free’.
Chronic renal failure patients
Haemoglobin concentration
In patients with chronic renal failure, maintenance haemoglobin concentration should not exceed the
upper limit of the target haemoglobin concentration recommended in section 4.2. In clinical trials, an
increased risk of death, serious cardiovascular events or cerebrovascular events including stroke were
observed when ESAs were administered to target a haemoglobin of greater than 12 g/dl (7.5 mmol/l).
Controlled clinical trials have not shown significant benefits attributable to the administration of
epoetins when haemoglobin concentration is increased beyond the level necessary to control
symptoms of anaemia and to avoid blood transfusion.
Haemoglobin levels should be measured on a regular basis until a stable level is achieved and
periodically thereafter. The rate of increase in haemoglobin should be approximately 1 g/dl
(0.62 mmol/l) per month and should not exceed 2 g/dl (1.25 mmol/l) per month to minimize the risk of
developing or worsening of hypertension.
Chronic renal failure patients treated with Retacrit by the subcutaneous route should be monitored
regularly for loss of efficacy, defined as absent or decreased response to Retacrit treatment in patients
who previously responded to such therapy. This is characterised by a sustained decrease in
haemoglobin despite an increase in Retacrit dosage.
36
Non response to erythropoietin therapy should prompt a search for causative factors. These include:
iron, folate, or Vitamin B
12
deficiency; aluminium intoxication; intercurrent infections; inflammatory
or traumatic episodes; occult blood loss; haemolysis, and bone marrow fibrosis of any origin.
Cases of antibody-mediated PRCA have been very rarely reported in chronic renal failure patients
with erythropoietin administered by the subcutaneous route. In patients developing sudden lack of
efficacy, defined by a decrease in haemoglobin (1-2 g/dl per month) with increased need for
transfusions, a reticulocyte count should be obtained and typical causes of non-response (e.g. iron,
folate, or Vitamin B
12
-deficiency, aluminium intoxication, infection or inflammation, blood loss, and
haemolysis) should be investigated. If no cause is identified, a bone marrow examination should be
considered for diagnosis of PRCA.
If PRCA is diagnosed, therapy with Retacrit must be immediately discontinued and testing for
erythropoietin antibodies should be considered. Patients should not be switched to another medicinal
product as anti-erythropoietin antibodies cross-react with other erythropoietins. Other causes of PRCA
should be excluded, and appropriate therapy initiated.
Monitoring of reticulocyte count on a regular basis is recommended to detect possible occurrence of
lack of efficacy in chronic renal failure patients.
Hyperkalaemia has been observed in isolated cases. In chronic renal failure patients, correction for
anaemia may lead to increased appetite, and potassium and protein intake. Dialysis prescriptions may
have to be adjusted periodically to maintain urea, creatinine and potassium in the desired range. Serum
electrolytes should be monitored in chronic renal failure patients. If an elevated (or rising) serum
potassium level is detected then consideration should be given to ceasing erythropoietin administration
until hyperkalaemia has been corrected.
An increase in heparin dose during haemodialysis is frequently required during the course of therapy
with erythropoietin as a result of the increased packed cell volume. Occlusion of the dialysis system is
possible if heparinisation is not optimum.
Based on information available to date, correction of anaemia with erythropoietin in adult patients
with renal insufficiency not yet undergoing dialysis does not accelerate the rate of progression of renal
insufficiency.
Adult cancer patients with symptomatic anaemia receiving chemotherapy
In cancer patients receiving chemotherapy, the 2-3 week delay between erythropoietin administration
and the appearance of erythropoietin-induced red cells should be taken into account when assessing if
Retacrit therapy is appropriate (patient at risk of being transfused).
Haemoglobin levels should be closely monitored until a stable level is achieved and periodically
thereafter. If the rate of increase in haemoglobin exceeds 2 g/dl (1.25 mmol/l) per month or the
haemoglobin level exceeds 12 g/dl (7,5 mmol/l), the dose adjustment detailed in section 4.2 should be
thoroughly performed to minimise the risk of thrombotic events (see section 4.2).
As an increased incidence of thrombotic vascular events (TVEs) has been observed in cancer patients
receiving erythropoietic agents (see section 4.8), this risk should be carefully weighed against the
benefit to be derived from treatment (with Retacrit) particularly in cancer patients with an increased
risk of thrombotic vascular events, such as obesity and patients with a prior history of TVEs (e.g. deep
venous thrombosis or pulmonary embolism).
Adult surgery patients in an autologous predonation programme
All special warnings and precautions associated with autologous predonation programs, especially
routine volume replacement, should be respected.
37
Tumour growth potential
Epoetins are growth factors that primarily stimulate red blood cell production. Erythropoietin
receptors may be expressed on the surface of a variety of tumour cells. As with all growth factors,
there is a concern that epoetins could stimulate the growth of any type of malignancy. In several
controlled studies, epoetins have not been shown to improve overall survival or decrease the risk of
tumour progression in patients with anaemia associated with cancer.
Several controlled clinical studies in which epoetins were administered to patients with a variety of
common tumours including squamous head and neck cancer, lung cancer, and breast cancer, have
shown an unexplained excess mortality.
In controlled clinical studies, use of Epoetin alfa and other erythropoiesis-stimulating agents (ESAs)
have shown:
•
shortened time to tumour progression in patients with advanced head and neck cancer
receiving radiation therapy when administered to target a haemoglobin of greater than 14 g/dl
(8.7 mmol/l),
•
shortened overall survival and increased deaths attributed to disease progression at 4 months
in patients with metastatic breast cancer receiving chemotherapy when administered to target a
haemoglobin of 12-14 g/dl (7.5 -8.7 mmol/l),
•
increased risk of death when administered to target a haemoglobin of 12 g/dl (7.5 mmol/l) in
patients with active malignant disease receiving neither chemotherapy nor radiation therapy.
ESAs are not indicated for use in this patient population.
In view of the above, in some clinical situations blood transfusion should be the preferred treatment
for the management of anaemia in patients with cancer. The decision to administer recombinant
erythropoietins should be based on a benefit-risk assessment with the participation of the individual
patient, which should take into account the specific clinical context. Factors that should be considered
in this assessment should include the type of tumour and its stage; the degree of anaemia; life-
expectancy; the environment in which the patient is being treated; and patient preference (see section
5.1).
4.5
There is no evidence to indicate that treatment with erythropoietin alters the metabolism of other
medicinal products.
However, since ciclosporin is bound by red blood cells there is potential for interactions with other
medicinal products. If erythropoietin is given concomitantly with ciclosporin, blood levels of
ciclosporin should be monitored and the dose of ciclosporin adjusted as the haematocrit rises.
No evidence exists that indicates an interaction between epoetin alfa and G-CSF or GM-CSF with
regard to haematological differentiation or proliferation of tumour biopsy specimens in vitro.
4.6
There are no adequate and well-controlled studies in pregnant women. Studies in animals have shown
reproduction toxicity (see section 5.3). Consequently, erythropoietin should generally be used during
pregnancy and lactation only if the potential benefit outweighs the potential risk to the foetus.
4.7
Retacrit has no or negligible influence on the ability to drive and use machines.
4.8
38
Retacrit is a biological medicinal product. Data from clinical studies with Retacrit are in line with the
safety profile of other authorized erythropoietins. Based on the results from clinical trials with other
authorized erythropoietins approximately 8% of patients treated with erythropoietin are expected to
predominantly in patients with chronic renal failure or underlying malignancies. These undesirable
effects are most commonly headache and a dose dependent increase in blood pressure. Hypertensive
crisis with encephalopathy-like symptoms can occur. Attention should be paid to sudden stabbing
migraine-like headaches as a possible warning signal.
Thrombotic/vascular events, such as myocardial ischaemia, myocardial infarction, cerebrovascular
accidents (cerebral haemorrhage and cerebral infarction), transient ischaemic attacks, deep vein
thrombosis, arterial thrombosis, pulmonary emboli, aneurysms, retinal thrombosis, and clotting of an
artificial kidney have been reported in patients receiving erythropoietic agents.
Antibody-mediated erythroblastopenia (PRCA) has been reported after months to years of treatment
with epoetin alfa. In most of these patients, antibodies to erythropoietins have been observed (see
sections 4.3 and 4.4).
In this section frequencies of undesirable effects are defined as follows: Very common (
>
1/10);
common (
>
1/100 to <1/10); uncommon (
>
1/1,000 to <1/100); rare (
>
1/10,000 to <1/1,000); very rare
(<1/10,000), not known (frequency cannot be estimated from the available data).
SOC
Frequency
ADR
very rare
Thrombocytosis (see section 4.4)
Blood and lymphatic system
disorders
Frequency not known
Antibody-mediated erythroblastopenia
(PRCA)
Immune system disorders
rare
Hypersensitivity reactions
very rare
anaphylactic reaction
very common
dizziness (chronic renal failure
patients)
headache (cancer patients)
Nervous system disorders
common
dizziness (cancer patients)
headache (chronic renal failure
patients)
stroke
uncommon
cerebral haemorrhage
Frequency not known
cerebral infarction
transient ischaemic attacks
hypertensive encephalopathy
Eye disorders
Frequency not known
retinal thrombosis
Cardiac disorders
Frequency not known
myocardial infarction
myocardial ischaemia
common
deep vein thrombosis (cancer patients)
increase in blood pressure
Vascular disorders
aneurysms
arterial thrombosis
deep vein thrombosis (chronic renal
failure patients)
hypertensive crisis
Frequency not known
Respiratory, thoracic and
mediastinal disorders
common
pulmonary embolism (cancer patients)
Frequency not known
pulmonary embolism (chronic renal
failure patients)
Skin and subcutaneous tissue
disorders
common
Non-specific skin rashes
very rare
Angioedema
39












































Frequency not known
pruritus
very common
joint pains (chronic renal failure
patients)
Musculoskeletal and
connective tissue disorders
common
joint pains (cancer patients)
very common
"Flu-like" symptoms (chronic renal
failure patients)
feelings of weakness (chronic renal
failure patients)
tiredness (chronic renal failure
patients)
General disorders and
administration site conditions
common
"Flu-like" symptoms (cancer patients)
feelings of weakness (cancer patients)
tiredness (cancer patients)
Injury, poisoning and
procedural complications
common
clotting of an artificial kidney
Adult and paediatric haemodialysis patients, adult peritoneal dialysis patients and adult patients with
renal insufficiency not yet undergoing dialysis
The most frequent adverse reaction during treatment with epoetin alfa is a dose-dependent increase in
blood pressure or aggravation of existing hypertension. These increases in blood pressure can be
treated with medicinal products. Moreover, monitoring of the blood pressure is recommended
particularly at the start of therapy. The following reactions have also occurred in isolated patients with
normal or low blood pressure: hypertensive crisis with encephalopathy-like symptoms (e.g. headaches
and confused state) and generalised tonoclonal seizures, requiring the immediate attention of a
physician and intensive medical care. Particular attention should be paid to sudden stabbing migraine
like headaches as a possible warning signal.
Shunt thromboses may occur, especially in patients who have a tendency to hypotension or whose
arteriovenous fistulae exhibit complications (e.g. stenoses, aneurysms, etc.). Early shunt revision and
thrombosis prophylaxis by administration of acetylsalicylic acid, for example, is recommended in
these patients.
Adult cancer patients with symptomatic anaemia receiving chemotherapy
Hypertension may occur in epoetin alfa treated patients. Consequently, haemoglobin and blood
pressure should be closely monitored.
An increased incidence of thrombotic vascular events (see section 4.4 and section 4.8 - General) has
been observed in patients receiving erythropoietic agents.
Surgery patients in autologous predonation programmes
Independent of erythropoietin treatment, thrombotic and vascular events may occur in surgical patients
with underlying cardiovascular disease following repeated phlebotomy. Therefore, routine volume
replacement should be performed in such patients.
4.9
The therapeutic margin of erythropoietin is very wide. Overdose of erythropoietin may produce effects
that are extensions of the pharmacological effects of the hormone. Phlebotomy may be performed if
excessively high haemoglobin levels occur. Additional supportive care should be provided as
necessary.
40




















5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Antianaemic preparations, erythropoietin
ATC code: B03XA01
Retacrit is a biosimilar medicinal product. Detailed information is available on the website of the
European Medicines Agency http://www.ema.europa.eu.
Erythropoietin is a glycoprotein that stimulates, as a mitosis-stimulating factor and differentiating
hormone, the formation of erythrocytes from precursors of the stem cell compartment.
The apparent molecular weight of erythropoietin is 32,000-40,000 Dalton. The protein moiety of the
molecule contributes about 58% of total molecular weight and consists of 165 amino acids. The four
carbohydrate chains are attached via three N-glycosidic bonds and one O-glycosidic bond to the
protein. Epoetin zeta is identical in its amino acid sequence and similar in carbohydrate composition to
endogenous human erythropoietin that has been isolated from the urine of anaemic patients.
The biological efficacy of erythropoietin has been demonstrated in various animal models
in vivo
(normal and anaemic rats, polycythaemic mice). After administration of erythropoietin, the number of
erythrocytes, the Hb values and reticulocyte counts increase as well as the
59
Fe-incorporation rate.
An increased
3
H-thymidine incorporation in the erythroid nucleated spleen cells has been found
in vitro
(mouse spleen cell culture) after incubation with erythropoietin. It could be shown with the aid
of cell cultures of human bone marrow cells that erythropoietin stimulates erythropoiesis specifically
and does not affect leucopoiesis. Cytotoxic actions of erythropoietin on bone marrow cells could not
be detected.
As with other haematopoietic growth factors, erythropoietin has shown
in vitro
stimulating properties
on human endothelial cells.
721 cancer patients receiving non-platinum chemotherapy were included in three placebo-controlled
studies, 389 patients with haematological malignancies (221 multiple myeloma, 144 non-Hodgkin's
lymphoma, and 24 other haematological malignancies) and 332 with solid tumours (172 breast,
64 gynaecological, 23 lung, 22 prostate, 21 gastrointestinal, and 30 other tumour types). In two large,
open-label studies, 2697 cancer patients receiving non-platinum chemotherapy were included,
1895 with solid tumours (683 breast, 260 lung, 174 gynaecological, 300 gastrointestinal, and 478 other
tumour types) and 802 with haematological malignancies.
In a prospective, randomised, double-blind, placebo-controlled trial conducted in 375 anaemic patients
with various non-myeloid malignancies receiving non-platinum chemotherapy, there was a significant
reduction of anaemia-related sequelae (e.g. fatigue, decreased energy, and activity reduction), as
measured by the following instruments and scales: Functional Assessment of Cancer
Therapy-Anaemia (FACT-An) general scale, FACT-An fatigue scale, and Cancer Linear Analogue
Scale (CLAS). Two other smaller, randomized, placebo-controlled trials failed to show a significant
improvement in quality of life parameters on the EORTC-QLQ-C30 scale or CLAS, respectively.
Erythropoietin is a growth factor that primarily stimulates red cell production. Erythropoietin receptors
may be expressed on the surface of a variety of tumour cells.
Survival and tumour progression have been examined in five large controlled studies involving a total
of 2833 patients, of which four were double-blind placebo-controlled studies and one was an open-
label study. The studies either recruited patients who were being treated with chemotherapy (two
studies) or used patient populations in which erythropoiesis stimulating agents are not indicated:
anaemia in patients with cancer not receiving chemotherapy, and head and neck cancer patients
41
receiving radiotherapy. The target haemoglobin concentration in two studies was > 13 g/dl; in the
remaining three studies it was 12-14 g/dl. In the open-label study there was no difference in overall
survival between patients treated with recombinant human erythropoietin and controls. In the four
placebo-controlled studies the hazard ratios for overall survival ranged between 1.25 and 2.47 in
favour of controls. These studies have shown a consistent unexplained statistically significant excess
mortality in patients who have anaemia associated with various common cancers who received
recombinant human erythropoietin compared to controls. Overall survival outcome in the trials could
not be statisfactorily explained by differences in the incidence of thrombosis and related complications
between those given recombinant human erythropoietin and those in the control group.
A systematic review has also been performed involving more than 9000 cancer patients participating
in 57 clinical trials. Meta-analysis of overall survival data produced a hazard ratio point estimate of
1.08 in favour of controls (95% CI: 0.99, 1,18; 42 trials and 8167 patients). An increased relative risk
of thromboembolic events (RR 1.67, 95% CI: 1.35, 2.06, 35 trials and 6769 patients) was observed in
patients treated with recombinant human erythropoietin. There is an increased risk for
thromboembolic events in patients with cancer treated with recombinant human erythropoietin and a
negative impact on overall survival cannot be excluded. The extent to which these outcomes might
apply to the administration of recombinant human erythropoietin to patients with cancer, treated with
chemotherapy to achieve haemoglobin concentrations less than 13 g/dl, is unclear because few patients
with these characteristics were included in the data reviewed.
A patient-level data analysis has also been performed on more than 13.900 cancer patients (chemo-,
radio-, chemoradio-, or no therapy) participating in 53 controlled clinical trials involving several
epoetins. Meta-analysis of overall survival data produced a hazard ratio point estimate of 1.06 in
favour of controls (95% CI: 1.00, 1.12; 53 trials and 13933 patients) and for the cancer patients
receiving chemotherapy, the overall survival hazard ratio was 1.04 (95% CI: 0.97, 1.11; 38 trials and
10.441 patients). Meta-analyses also indicate consistently a significantly increased relative risk of
thromboembolic events in cancer patients receiving recombinant human erythropoietin (see section
4.4).
In a randomised, double-blind, placebo-controlled study of 4,038 CRF patients not on dialysis with
type 2 diabetes and haemoglobin levels ≤ 11 g/dL, patients received either treatment with darbepoetin
alfa to target haemoglobin levels of 13 g/dL or placebo (see section 4.4). The study did not meet either
primary objective of demonstrating a reduction in risk for all-cause mortality, cardiovascular
morbidity, or end stage renal disease (ESRD). Analysis of the individual components of the composite
endpoints showed the following HR (95% CI): death 1.05 (0.92, 1.21), stroke 1.92 (1.38, 2.68),
congestive heart failure (CHF) 0.89 (0.74, 1.08), myocardial infarction (MI) 0.96 (0.75, 1.23),
hospitalisation for myocardial ischaemia 0.84 (0.55, 1.27), ESRD 1.02 (0.87, 1.18).
5.2
Pharmacokinetic properties
Measurement of erythropoietin following multiple dose intravenous administration revealed a half-life
of approximately 4 hours in healthy volunteers and a somewhat more prolonged half-life of
approximately 5 hours in renal failure patients. A half-life of approximately 6 hours has been reported
in children.
Subcutaneous route
Following subcutaneous injection, serum levels of erythropoietin are much lower than the levels
achieved following intravenous injection, the levels increase slowly and reach a peak between 12 and
18 hours postdose. The peak is always well below the peak achieved using the intravenous route
(approximately 1/20th of the value).
There is no accumulation: the levels remain the same, whether they are determined 24 hours after the
first injection or 24 hours after the last injection.
42
Intravenous route
The half-life is difficult to evaluate for the subcutaneous route and is estimated to be about 24 hours.
The bioavailability of subcutaneous injectable erythropoietin is much lower than that of the
intravenous medicinal product and is approximately 20%.
5.3
Preclinical safety data
In some pre-clinical toxicological studies in dogs and rats, but not in monkeys, erythropoietin therapy
was associated with subclinical bone marrow fibrosis (bone marrow fibrosis is a known complication
of chronic renal failure in humans and may be related to secondary hyperparathyroidism or unknown
factors. The incidence of bone marrow fibrosis was not increased in a study of haemodialysis patients
who were treated with erythropoietin for 3 years compared to a matched control group of dialysis
patients who had not been treated with erythropoietin).
In animal studies, erythropoietin has been shown to decrease foetal body weight, delay ossification
and increase foetal mortality when given in weekly doses of approximately 20 times the recommended
human weekly dose. These changes are interpreted as being secondary to decreased maternal body
weight gain.
Erythropoietin did not show any changes in bacterial and mammalian cell culture mutagenicity tests
and an
in vivo
micronucleus test in mice. Long-term carcinogenicity studies have not been carried out.
There are conflicting reports in the literature regarding whether erythropoietin may play a major role
as tumour proliferator. These reports are based on
in vitro
findings from human tumour samples, but
are of uncertain significance in the clinical situation.
6.
6.1
List of Excipients
Disodium phosphate dihydrate
Sodium dihydrogen phosphate dihydrate
Sodium chloride
Calcium chloride dihydrate
Polysorbate 20
Glycine
Leucine
Isoleucine
Threonine
Glutamic acid
Phenylalanine
Water for injections
Sodium hydroxide (pH adjuster)
Hydrochloric acid (pH adjuster)
6.2
Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal
products.
6.3
Shelf life
2 years
6.4
Special precautions for storage
Store in a refrigerator (2°C - 8°C). Do not freeze.
Keep the pre-filled syringe in the outer carton in order to protect from light.
43
For the purpose of ambulatory use, the patient may remove the product from the refrigerator and store
it at room temperature (not above 25°C) for one single period of up to 3 days.
6.5
Nature and contents of container
Pre-filled syringe Type I glass with a fixed steel injection needle and a plunger stopper with PTFE
coating.
Each pre-filled syringe contains 0.9 ml solution for injection.
Each pack contains 1 or 6 pre-filled syringes.
Not all pack sizes may be marketed.
6.6
Special precautions for disposal and other handling
Handling instructions for Retacrit:
1.
After removing one syringe from the blister pack the solution should be checked to ensure that
it is clear, colourless and practically free from visible particles.
2.
The protective cap is removed from the injection needle and air is expelled from the syringe and
needle by holding the syringe vertically and gently pressing the plunger upwards.
3.
The syringe is now ready for use.
Retacrit must not be used if
•
The blister sealing is broken or the blister is damaged in any way.
•
The liquid is coloured or you can see particles floating in it.
•
Any liquid has leaked out of the pre-filled syringe or condensation is visible within the sealed
blister.
•
It may have been accidentally frozen.
This medicinal product is for single use only.
Do not shake.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Hospira UK Limited
Queensway
Royal Leamington Spa
Warwickshire
CV31 3RW
United Kingdom
8.
EU/1/07/431/005
EU/1/07/431/006
9.
18/12/2007
44
1.
FURTHER INFORMATION
What Retacrit contains
–
The active substance is epoetin zeta (produced by recombinant DNA technology in Chinese
Hamster Ovary (CHO) cell line.
Retacrit 1000 IU/0.3ml solution for injection in pre-filled syringe
1 pre-filled syringe with 0.3 ml solution for injection contains 1000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 3333 IU Epoetin zeta
per ml.
Retacrit 2000 IU/0.6ml solution for injection in pre-filled syringe
1 pre-filled syringe with 0.6 ml solution for injection contains 2000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 3333 IU Epoetin zeta
per ml.
1 pre-filled syringe with 0.9 ml solution for injection contains 3000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 3333 IU Epoetin zeta
per ml.
Retacrit 4000 IU/0.4ml solution for injection in pre-filled syringe
209
Retacrit 3000 IU/0.9ml solution for injection in pre-filled syringe
1 pre-filled syringe with 0.4 ml solution for injection contains 4000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 10000 IU Epoetin zeta
per ml.
1 pre-filled syringe with 0.5 ml solution for injection contains 5000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 10000 IU Epoetin zeta
per ml.
Retacrit 6000 IU/0.6ml solution for injection in pre-filled syringe
1 pre-filled syringe with 0.6 ml solution for injection contains 6000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 10000 IU Epoetin zeta
per ml.
Retacrit 8000 IU/0.8ml solution for injection in pre-filled syringe
1 pre-filled syringe with 0.8 ml solution for injection contains 8000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 10000 IU Epoetin zeta
per ml.
1 pre-filled syringe with 1.0 ml solution for injection contains 10000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 10000 IU Epoetin zeta
per ml.
Retacrit 20000 IU/0.5ml solution for injection in pre-filled syringe
1 pre-filled syringe with 0.5 ml solution for injection contains 20000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 40000 IU Epoetin zeta
per ml.
Retacrit 30000 IU/0.75ml solution for injection in pre-filled syringe
1 pre-filled syringe with 0.75 ml solution for injection contains 30000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 40000 IU Epoetin zeta
per ml.
Retacrit 40000 IU/1.0ml solution for injection in pre-filled syringe
1 pre-filled syringe with 1.0 ml solution for injection contains 40000 international units (IU)
epoetin zeta (recombinant human erythropoietin). The solution contains 40000 IU Epoetin zeta
per ml.
The other ingredients are disodium phosphate dihydrate, sodium dihydrogen phosphate dihydrate,
sodium chloride, calcium chloride dihydrate, polysorbate 20, glycine, leucine, isoleucine, threonine,
glutamic acid, phenylalanine, water for injections, sodium hydroxide (pH adjuster), hydrochloric acid
(pH adjuster).
What Retacrit looks like and contents of the pack
Retacrit is presented as a clear and colourless solution for injection in a pre-filled syringe with a fixed
injection needle.
The pre-filled syringes contain between 0.3 and 1.0 ml solution, depending on the content of epoetin
zeta (see “What Retacrit contains”).
One pack contains 1, 4 or 6 pre-filled syringes.
Marketing Authorisation Holder
Hospira UK Limited
210
Retacrit 5000 IU/0.5ml solution for injection in pre-filled syringe
Retacrit 10000 IU/1.0ml solution for injection in pre-filled syringe
Queensway
Royal Leamington Spa
Warwickshire
CV31 3RW
United Kingdom
Manufacturer
STADA Arzneimittel AG
Stadastrasse 2-18
D-61118 Bad Vilbel
Germany
HOSPIRA Enterprises B.V.
Randstad 22-11
1316 BN Almere
The Netherlands
For any information about this medicine, please contact the local representative of the Marketing
Authorisation Holder:
België/Belgique/Belgien
Hospira Benelux BVBA
Tél/Tel: + 32 2 332 03 15
Luxembourg/Luxemburg
Hospira Benelux BVBA
Tél/Tel: + 32 2 332 03 15
България
Hospira UK Limited
Teл.: + 44 (0) 1926 820820
Magyarország
Pharmacenter Hungary Kft.
Tel.: + 36-1-209-5927
Česká republika
Movianto Česká republika s.r.o
Tel: + 420 548 134 400
Malta
Hospira UK Limited
Tel: + 44 (0) 1926 820820
Danmark
Hospira Nordic AB
Tlf: + 46 (0)8 672 85 00
Nederland
Hospira Benelux BVBA
Tel: + 32 2 332 03 15
Deutschland
Hospira Deutschland GmbH
Tel: + 49 (0) 89 43 77 77 0
Norge
Hospira Nordic AB
Tlf: + 46 (0)8 672 85 00
Eesti
Berren Medical, c/o Axellus OÜ
Tel: + 372 662 3573
Österreich
Astro-Pharma Vertrieb und Handel von
pharmazeutischen Produkten GmbH
Tel: + 43 (0)1 961 93 13
Ελλάδα
Aenorasis S.A.
Τηλ: + 30 210 6136332
Polska
Zaklady Farmaceutyczne S.A.
Tel.: + 481 26178048
España
Hospira
Productos Farmacéuticos y Hospitalarios S.L.
Tel: + 34 914847100
Portugal
Hospira Portugal Lda
Tel: + 351 214857434
France
România
211
Hospira France
Tél: + 33 (0) 826 30 03 02
Hospira UK Limited
Tel: + 44 (0) 1926 820820
Ireland
Hospira Ireland Limited
Tel: + 353 (0) 1 2962102
Slovenija
Valentis Pharmaceuticals d.o.0
Tel: + 386 1 2000603
Ísland
Hospira Nordic AB
Sími: + 46 (0)8 672 85 00
Slovenská republika
Pliva s.r.o
Tel: + 421 2 2060 2852
Italia
Hospira Italia Srl
Tel: + 39 0812405912
Suomi/Finland
Hospira Nordic AB
Puh/Tel: + 46 (0)8 672 85 00
Κύπρος
Hospira UK Limited
Τηλ: + 44 (0) 1926 820820
Sverige
Hospira Nordic AB
Tel: + 46 (0)8 672 85 00
Latvija
Berren Medical, c/o Axellus SIA
Tel: + 371 721 1629
United Kingdom
Hospira UK Limited
Tel: + 44 (0) 1926 820820
Lietuva
Berren Medical, c/o Axellus UAB
Tel: + 370 5 231 0654
This leaflet was last approved in
{MM/YYYY}.
Detailed information on this medicine is available on the European Medicines Agency web site:
http://www.ema.europa.eu.
212
Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/retacrit.html
Copyright © 1995-2021 ITA all rights reserved.