










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Sebivo 600 mg film-coated tablets
2.
Each film-coated tablet contains 600 mg telbivudine.
For a full list of excipients, see section 6.1.
3.
Film-coated tablet
White to slightly yellowish, oval film-coated tablet, imprinted with “LDT” on one side.
4.
Sebivo is indicated for the treatment of chronic hepatitis B in adult patients with compensated liver
disease and evidence of viral replication, persistently elevated serum alanine aminotransferase (ALT)
levels and histological evidence of active inflammation and/or fibrosis. See section 5.1 for details of
the study and specific patient characteristics on which this indication is based.
Therapy must be initiated by a physician experienced in the management of chronic hepatitis B
infection.
Adults
The recommended dose of Sebivo is 600 mg (one tablet) once daily, taken orally, with or without
food.
Sebivo oral solution may be considered for patients who have difficulties swallowing tablets.
Duration of therapy
The optimal treatment duration is unknown. Treatment discontinuation should be considered as
follows:
In HBeAg-positive patients without cirrhosis, treatment should be administered for at least
6-12 months after HBe seroconversion (HBeAg loss and HBV DNA loss with anti-HBe
detection) is confirmed or until HBs seroconversion or there is evidence of loss of efficacy.
Serum ALT and HBV DNA levels should be followed regularly after treatment discontinuation
to detect any late virological relapse.
In HBeAg-negative patients without cirrhosis, treatment should be administered at least until
HBs seroconversion or until there is evidence of loss of efficacy. With prolonged treatment for
more than 2 years, regular reassessment is recommended to confirm that continuation of the
selected therapy remains appropriate for the patient.
On-treatment response at week 24 has been shown to be predictive of longer-term response (see
Table 7 in section 5.1) and might be useful for driving the management of patients treated with
telbivudine monotherapy.
2
Renal impairment
No adjustment of the recommended dose of telbivudine is necessary in patients whose creatinine
clearance is 50 ml/min. Adjustment of the dose is required in patients with creatinine clearance
< 50 ml/min, including those with end-stage renal disease (ESRD) on haemodialysis. A reduction of
the daily dose using Sebivo oral solution, as detailed in Table 1 below, is recommended. If use of the
oral solution is not possible, Sebivo film-coated tablets could be used as an alternative and dosing
should be adjusted by increasing the time interval between doses, as detailed in Table 1.
Table 1
Dosing regimen adjustment of Sebivo in patients with renal impairment
Creatinine clearance
(ml/min)
Telbivudine 20 mg/ml oral
solution
Daily dose adjustment
Telbivudine 600 mg film-coated
tablet
Alternative** dose adjustment with
increased dose intervals
50
600 mg (30 ml) once daily
600 mg once daily
30-49
400 mg (20 ml) once daily
600 mg once every 48 hours
< 30 (not requiring
dialysis)
200 mg (10 ml) once daily
600 mg once every 72 hours
ESRD*
120 mg (6 ml) once daily
600 mg once every 96 hours
* End stage renal disease
** In case use of the oral solution is not possible
The proposed dose modifications are based on extrapolation and may not be optimal. The safety and
effectiveness of these dosing adjustment guidelines have not been clinically evaluated. Therefore,
close clinical monitoring is recommended in these patients.
End-stage renal disease patients
For patients with ESRD, Sebivo should be administered after haemodialysis (see section 5.2).
Hepatic impairment
No adjustment to the recommended dose of Sebivo is necessary in patients with hepatic impairment
(see section 5.2).
Children and adolescents
Sebivo is not recommended for use in children and adolescents below 16 years of age due to a lack of
data on safety and efficacy.
Elderly patients (age above 65 years)
No data are available to support a specific dose recommendation for patients over the age of 65 years
(see section 4.4).
Hypersensitivity to the active substance or to any of the excipients.
Combination of telbivudine with pegylated or standard interferon alfa (see sections 4.4 and 4.5).
Severe acute exacerbations of chronic hepatitis B are relatively frequent, and are characterised by
transient elevation of serum ALT. Following initiation of antiviral treatment, serum ALT may rise in
some patients while serum levels of HBV DNA fall (see section 4.8). On average, 4-5 weeks elapsed
prior to the occurrence of an exacerbation in patients treated with telbivudine. Overall, ALT flares
occurred more frequently in HBeAg-positive patients than in HBeAg-negative patients. In patients
with compensated liver disease, this elevation of serum ALT is generally not accompanied by elevated
levels of serum bilirubin or by other signs of hepatic decompensation. The risk of hepatic
3




decompensation – and of a subsequent exacerbation of hepatitis – may be elevated in patients with
cirrhosis. Such patients should therefore be closely monitored.
Exacerbations of hepatitis have also been reported in patients who have terminated treatment of
hepatitis B. Post-treatment ALT flares are normally associated with increases in serum HBV DNA
levels, and the majority of such cases have proven to be self-limiting. Nonetheless, there have also
been reports of severe – and sometimes fatal – post-treatment disease exacerbations. Therefore,
hepatic function should be monitored at regular intervals with both clinical and laboratory follow-up
for at least 6 months after discontinuation of hepatitis B therapy.
Occurrence of lactic acidosis (in the absence of hypoxaemia) sometimes fatal, and usually associated
with severe hepatomegaly with steatosis have been reported with the use of nucleoside/nucleotide
analogues. As telbivudine is a nucleoside analogue, this risk cannot be excluded. Treatment with
nucleoside analogues should be discontinued when rapidly elevating aminotransferase levels,
progressive hepatomegaly or metabolic/lactic acidosis of unknown aetiology occur. Benign digestive
symptoms, such as nausea, vomiting and abdominal pain, may be indicative of lactic acidosis
development. Severe cases, sometimes with fatal outcome, were associated with pancreatitis, liver
failure/hepatic steatosis, renal failure and higher levels of serum lactate. Caution should be exercised
when prescribing nucleoside analogues to any patient (particularly obese women) with hepatomegaly,
hepatitis or other known risk factors for liver disease. These patients should be followed closely.
Muscular effects
Cases of myopathy and myalgia have been reported with telbivudine use several weeks to months after
starting therapy (see section 4.8). Cases of rhabdomyolysis have been reported during post-marketing
use of telbivudine (see section 4.8).
Myopathy, defined as persistent unexplained muscle aches and/or muscle weakness regardless of the
degree of increases in creatine kinase levels, should be considered in any patient with diffuse
unexplained myalgias, muscle tenderness, muscle weakness or myositis (defined as myopathy with
histological evidence of muscle damage). Patients should be advised to report promptly any persistent
unexplained muscle aches, pain, tenderness or weakness. If any of these symptoms are reported, a
detailed muscle examination should be performed in order to evaluate muscle function. Telbivudine
therapy should be discontinued if myopathy is diagnosed.
It is not known whether the risk of myopathy during treatment with telbivudine is increased with
concurrent administration of other medicinal products associated with myopathy (e.g. statins, fibrates,
or ciclosporin). Physicians considering concomitant treatment with other agents associated with
myopathy should weigh carefully the potential benefits and risks and should monitor patients for any
signs or symptoms suggestive of myopathy.
Peripheral neuropathy
Peripheral neuropathy has been uncommonly reported in telbivudine-treated patients. If peripheral
neuropathy is suspected, treatment with telbivudine should be reconsidered (see section 4.8).
An increased risk of developing peripheral neuropathy has been observed in one study when
telbivudine and pegylated interferon alfa-2a were co-administered (see section 4.5). Such increased
risk cannot be excluded for other interferon alfa (pegylated or standard). Moreover, the benefit of the
combination of telbivudine with interferon alfa (pegylated or standard) is not currently established.
Therefore, the combination of telbivudine with pegylated or standard interferon alfa is contraindicated
(see section 4.3).
Renal function
Telbivudine is eliminated primarily by renal excretion,
therefore dose interval adjustment is
recommended in patients with creatinine clearance < 50 ml/min, including patients on haemodialysis.
The effectiveness of dosing interval adjustment has not been clinically evaluated. Therefore,
virological response should be closely monitored in patients with increased dosage interval (see
sections 4.2 and 5.2).
4
Patients with cirrhosis without decompensation
Due to the limited data available (about 3% of patients enrolled had cirrhosis), telbivudine should be
used with particular caution in cirrhotic patients. These patients should be closely monitored for
clinical, biochemical and virological parameters associated with hepatitis B during treatment and after
treatment is discontinued.
Patients with cirrhosis with decompensation
There are no efficacy and safety data in patients with decompensated cirrhosis.
Patients with previous exposure to nucleoside/nucleotide analogs
In vitro
, telbivudine was not active against the HBV strains containing rtM204V/rtL180M or rtM204I
mutations (see section 5.1). Telbivudine monotherapy is not an option for patients with established
lamivudine-resistant hepatitis B virus infection. Patients who failed to achieve virological response
following treatment with lamivudine for more than 24 weeks are unlikely to benefit from telbivudine
monotherapy. There is currently no clinical data to properly assess the benefit and risk of switching to
telbivudine for lamivudine-treated patients who achieve complete viral suppression on lamivudine.
There are no data on telbivudine treatment in patients with established adefovir-resistant hepatitis B
virus single mutations of rtN236T or A181V. Results from cell-based assays showed that the adefovir
resistance-associated substitution A181V had 1.5- to approximately 4-fold reduced susceptibility to
telbivudine.
Liver transplant recipients
The safety and efficacy of telbivudine in liver transplant recipients are unknown.
Elderly patients
Clinical studies of telbivudine did not include sufficient numbers of patients 65 years of age to
determine whether they respond differently from younger subjects. In general, caution must be
exercised when prescribing Sebivo to elderly patients in view of the greater frequency of decreased
renal function due to concomitant disease or concomitant use of other medicinal products.
Other special populations
Sebivo has not been investigated in co-infected hepatitis B patients (e.g. patients co-infected with
human immunodeficiency virus [HIV], hepatitis C virus [HCV] or hepatitis D virus [HDV]).
General
Patients should be advised that treatment with Sebivo has not been shown to reduce the risk of
transmission of HBV to others through sexual contact or blood contamination.
Telbivudine is not recommended to be used with lamivudine because in a phase II study, the treatment
response observed with combination therapy of telbivudine and lamivudine was lower than with
telbivudine alone.
There are currently no efficacy and safety data for other antiviral combinations with telbivudine.
Since telbivudine is eliminated primarily by renal excretion, co-administration of Sebivo with
substances that affect renal function (such as aminoglycosides, loop diuretics, platinum compounds,
vancomycin, amphotericin B) may affect plasma concentrations of telbivudine and/or the co-
administered substance. The combination of telbivudine with these medicinal products should be used
with caution. The steady-state pharmacokinetics of telbivudine were unaltered following multiple dose
administration in combination with lamivudine, adefovir dipivoxil, tenofovir disoproxil fumarate,
ciclosporin or pegylated interferon alfa-2a. In addition, telbivudine does not alter the pharmacokinetics
of lamivudine, adefovir dipivoxil, tenofovir disoproxil fumarate or ciclosporin. No definitive
conclusion could be drawn regarding the effects of telbivudine on the pharmacokinetics of pegylated
5
interferon due to high interindividual variability of pegylated interferon alfa-2a concentrations. A
clinical trial investigating the combination of telbivudine, 600 mg daily, with pegylated interferon
alfa-2a, 180 micrograms once weekly by subcutaneous administration, indicates that this combination
is associated with an increased risk of developing peripheral neuropathy. The mechanism behind these
events is not known (see section 4.4). The combination of telbivudine with any interferon alfa-
containing product is contraindicated (see section 4.3).
Telbivudine is not a substrate, inhibitor or inducer of the cytochrome P450 (CYP450) enzyme system
(see section 5.2).Therefore, the potential for CYP450-mediated drug interactions involving Sebivo is
low.
For telbivudine no clinical data on exposed pregnancies are available. Animal studies do not indicate
direct harmful effects with respect to pregnancy, embryonal/foetal development, parturition or
postnatal development (see section 5.3). Studies in pregnant rats and rabbits showed that telbivudine
crosses the placenta. Studies in pregnant rabbits showed early delivery and/or abortion secondary to
maternal toxicity. Sebivo should be used during pregnancy only if the benefit to the mother outweighs
the potential risk to the foetus.
There are no data on the effect of telbivudine on transmission of HBV from mother to infant.
Therefore, appropriate interventions should be used to prevent neonatal acquisition of HBV infection.
Telbivudine is excreted in the milk of rats. It is not known whether telbivudine is excreted in human
milk. Women should not breast-feed if they are taking Sebivo.
There are no clinical data on the effects of telbivudine on male or female fertility. In reproductive
toxicology studies in adult animals, fertility was slightly reduced when both male and female rats
received telbivudine. The adverse effects on fertility were greater in a separate study in juvenile
animals when both sexes received telbivudine (see section 5.3).
No studies on the effects on the ability to drive and use machines have been performed.
Assessment of adverse reactions is mainly based on two studies (NV-02B-007 “GLOBE” and NV-
02B-015) in which 1,699 patients with chronic hepatitis B received double-blind treatment with
telbivudine 600 mg/day (n = 847) or lamivudine (n = 852) for 104 weeks.
In the 104-week clinical studies, reported adverse reactions were usually classified as mild or
moderate in severity. The most common adverse events with at least a possible relation to telbivudine
were grade 3/4 blood creatine kinase elevations (6.8%), fatigue (4.4%), headache (3.0%) and nausea
(2.6%).
6
Table 2 lists the adverse reactions according to MedDRA system organ class and frequency using the
following convention: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to
<1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from
the available data). Within each frequency grouping, adverse reactions are presented in order of
decreasing seriousness.
Table 2
Adverse reactions
Metabolism and nutrition disorders
Not known*
Lactic acidosis as a secondary event often
associated with serious conditions (e.g. multi-
organ failure or sepsis)
Nervous system disorders
Common
Dizziness, headache
Uncommon
Peripheral neuropathy, dysgeusia, hypoaesthesia,
paresthesia, sciatica
Respiratory, thoracic and mediastinal
disorders
Common
Cough
Gastrointestinal disorders
Common
Blood amylase increased, diarrhoea, blood lipase
increased, nausea, abdominal pain
Hepatobiliary disorders
Common
Blood alanine aminotransferase increased
Uncommon
Aspartate aminotransferase increased
Skin and subcutaneous tissue disorders
Common
Rash
Musculoskeletal and connective tissue
disorders
Common
Blood creatine phosphokinase increased
Uncommon
Arthralgia, myalgia, myopathy/myositis, pain in
the extremities, back pain, muscle spasm, neck
pain, flank pain
Not known*
Rhabdomyolysis
General disorders and administration site
conditions
Common
Fatigue
Uncommon
Malaise
*
Based on post-marketing reports. Since these reports are from a population of uncertain size it is
not possible to reliably estimate their frequency and therefore the frequency is classed as “not
known”.
Creatine kinase
In the pooled analysis, by 104 weeks of treatment Grade 3/4 creatine kinase elevations (> 7x ULN)
occurred in 12.6% of telbivudine-treated patients and 4.0% of lamivudine-treated patients. Most
creatine kinase elevations were asymptomatic and creatine kinase values typically decreased by the
next visit on continued treatment. In the pivotal study NV-02B-007 (GLOBE), higher pre-treatment
CK values and Caucasian race were identified in both treatment groups as predictive factors for
Grade 3/4 elevations by 104 weeks (see section 4.4).
7
























ALT flares
The incidence of on treatment alanine aminotransferase (ALT) flares in the two treatment arms
according to AASLD (American Association for the Study of Liver Diseases) definition (ALT
elevation > 2x baseline and > 10x ULN) are further described in Table 3 below.
Table 3
Summary of on treatment ALT (IU/L) flare – Pooled NV-02B-007/NV-02B-015
ALT flare:
ALT elevation > 2x baseline and
> 10x ULN
Lamivudine
n/N (%)
Telbivudine
n/N (%)
Overall
67/852 (7.9)
41/847 (4.8)
Baseline to week 24
25/852 (2.9)
25/847 (3.0)
Week 24 to end of study
44/837 (5.3)
17/834 (2.0)
Periodic monitoring of hepatic function is recommended during treatment (see section 4.4).
Exacerbations of hepatitis B after discontinuation of treatment
Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued anti-
hepatitis B therapy including telbivudine (see section 4.4).
The incidence of post-treatment alanine aminotransferase (ALT) flares in the two treatment arms are
further described in Table 4 below.
Table 4
Summary of post-treatment ALT flares – Pooled NV-02B-007/NV-02B-015
Lamivudine
Telbivudine
ALT flare
n/N (%)
n/N (%)
ALT elevation > 2x baseline and > 10x ULN
10/180 (5.6)
9/154 (5.8)
There is no information on intentional overdose of telbivudine, but one subject was given an
unintentional overdose which was asymptomatic. Tested doses up to 1,800 mg/day, three times greater
than the recommended daily dose, have been well tolerated. A maximum tolerated dose of telbivudine
has not been determined. In the event of an overdose, Sebivo should be discontinued and appropriate
general supportive treatment applied as necessary.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Nucleoside and nucleotide reverse transcriptase inhibitors, ATC code:
J05AF11
Telbivudine is a synthetic thymidine nucleoside analogue with activity against HBV DNA
polymerase. It is efficiently phosphorylated by cellular kinases to the active triphosphate form, which
has an intracellular half-life of 14 hours. Telbivudine-5'-triphosphate inhibits HBV DNA polymerase
(reverse transcriptase) by competing with the natural substrate, thymidine 5'-triphosphate.
Incorporation of telbivudine-5'-triphosphate into viral DNA causes DNA chain termination, resulting
in inhibition of HBV replication. Telbivudine is an inhibitor of both HBV first strand
(EC
50
= 0.4-1.3 M) and second strand (EC
50
= 0.12-0.24 M) synthesis, and shows a distinct
preference for inhibiting second strand production. By contrast, telbivudine-5'-triphosphate at
8
















concentrations up to 100 M did not inhibit cellular DNA polymerases , , or . In assays relating to
mitochondrial structure, function and DNA content, telbivudine lacked appreciable toxic effect at
concentrations up to at 10 M and did not increase lactic acid production
in vitro
.
The
in vitro
antiviral activity of telbivudine was assessed in the HBV-expressing human hepatoma cell
line 2.2.15. The concentration of telbivudine that effectively inhibited 50% of viral synthesis (EC
50
)
was approximately 0.2 M. The antiviral activity of telbivudine is specific to the hepatitis B virus and
related hepadnaviruses. Telbivudine was not active against HIV
in vitro
. The absence of activity of
telbivudine against HIV has not been evaluated in clinical trials.
Clinical experience
The safety and efficacy of long-term (104 weeks) Sebivo treatment were evaluated in two active-
controlled clinical studies that included 1,699 patients with chronic hepatitis B (NV-02B-007 GLOBE
and NV-02B-015).
NV-02B-007 GLOBE study
The NV-02B-007 GLOBE study is a randomised, double-blind, multinational phase III study of
telbivudine compared to lamivudine for a treatment period of 104 weeks in 1,367 nucleoside-naïve
chronic hepatitis B HBeAg-positive and HBeAg-negative patients. The majority of the population
enrolled was Asian. The most common HBV genotypes were B (26%) and C (51%). A small number
(total of 98) of Caucasian patients were treated with telbivudine. The primary data analysis was
conducted after all patients had reached week 52.
HBeAg-positive patients
: The mean age of patients was 32 years, 74% were male, 82% were Asian,
12% were Caucasian, and 6% had previously received alfa-interferon therapy.
HBeAg-negative patients
: The mean age of patients was 43 years, 79% were male, 65% were Asian,
23% were Caucasian, and 11% had previously received alfa-interferon therapy.
Clinical results at week 52
Clinical and virological efficacy endpoints were evaluated separately in the HBeAg-positive and
HBeAg-negative patient populations. The primary endpoint of therapeutic response was a composite
serological endpoint requiring suppression of HBV DNA to < 5 log
10
copies/ml in conjunction with
either loss of serum HBeAg or ALT normalised. Secondary endpoints included histological response,
ALT normalisation, and various measures of antiviral efficacy.
Regardless of baseline characteristics, the majority of patients taking Sebivo showed histological,
virological, biochemical, and serological responses to treatment. Baseline ALT levels > 2 x ULN and
baseline HBV DNA < 9 log
10
copies/ml were associated with higher rates of eAg seroconversion in
HBeAg-positive patients. Patients who achieve HBV DNA levels < 3 log
10
copies/ml by week 24 had
optimal responses to treatment; conversely patients with HBV DNA levels > 4 log
10
copies/ml at
24 weeks had less favourable outcomes at week 52.
In HBeAg-positive patients, telbivudine was superior to lamivudine in therapeutic response (75.3% vs
67.0% responders; p = 0.0047). In HBeAg-negative patients, telbivudine was non-inferior to
lamivudine (75.2% and 77.2% responders; p = 0.6187). Caucasian ethnicity was associated with lower
treatment response to both antiviral agents used in the GLOBE trial; however the Caucasian patient
population was very limited (n = 98).
At week 24, 203 HBeAg-positive and 177 HBeAg-negative subjects achieved non-detectable HBV
DNA levels. Of those HBeAg positive subjects, 95% achieved non-detectable HBV DNA, 39%
achieved HBeAg seroconversion, 90% achieved ALT normalisation at week 52 and 0.5% exhibited
resistance at week 48. Similarly of those HBeAg-negative subjects, 96% achieved non-detectable
HBV DNA, 79% achieved ALT normalisation at week 52 and 0% exhibited resistance at week 48.
9
Selected virological, biochemical and serological outcome measures are shown in Table 5 and
histological response in Table 6.
Table 5 Virological, biochemical and serological endpoints at week 52 (NV-02B-007 GLOBE
study)
HBeAg-positive (n = 921)
HBeAg-negative (n = 446)
Response parameter
Telbivudine
600 mg
(n = 458)
Lamivudine
100 mg
(n = 463)
Telbivudine
600 mg
(n = 222)
Lamivudine
100 mg
(n = 224)
Mean HBV DNA
reduction from
baseline (log
10
copies/ml) ± SEM
1,2,3
-6.45 (0.11)
*
-5.54 (0.11)
-5.23 (0.13)
*
-4.40 (0.13)
% Patients HBV
DNA negative by
PCR
60%*
40%
88%*
71%
ALT normalisation
4
77%
75%
74%
79%
HBeAg
seroconversion
4
23%
22%
-
-
HBeAg loss
5
26%
23%
-
-
1
SEM: Standard error of mean
2
Roche COBAS Amplicor
®
PCR Assay (lower limit of quantification 300 copies/ml).
3
HBeAg-positive n = 443 and 444, HBeAg-negative n = 219 and 219, for both telbivudine and
lamivudine groups, respectively. The difference in populations is due to patient discontinuation from
the study and missing HBV DNA assessment at week 52.
4
HBeAg-positive n = 440 and 446, HBeAg-negative n = 203 and 207, for telbivudine and lamivudine
groups, respectively. ALT normalisation assessed only in patients with ALT > ULN at baseline.
5
n = 432 and 442, for telbivudine and lamivudine groups, respectively. HBeAg seroconversion and
loss assessed only in patients with detectable HBeAg at baseline.
*p < 0.0001
Table 6 Histological improvement and change in Ishak Fibrosis Score at week 52 (NV-02B-
007 GLOBE study)
HBeAg-positive (n = 921)
HBeAg-negative (n = 446)
Telbivudine
600 mg
(n = 384)
1
Lamivudine
100 mg
(n = 386)
1
Telbivudine
600 mg
(n = 199)
1
Lamivudine
100 mg
(n = 207)
1
Histological response
2
Improvement
71%*
61%
71%
70%
No improvement
17%
24%
21%
24%
Ishak Fibrosis Score
3
Improvement 42% 47% 49% 45%
No change 39% 32% 34% 43%
Worsening 8% 7% 9% 5%
Missing week 52 biopsy
12% 15% 9% 7%
1
Patients with ≥ one dose of study drug with evaluable baseline liver biopsies and baseline
Knodell Histological Activity Index (HAI) score > 3.
2
Histological response defined as a ≥ 2 point decrease in Knodell Necroinflammatory Score from
baseline with no worsening of the Knodell Fibrosis Score.
3
For Ishak Fibrosis Score, improvement measured as ≥ 1 point reduction in Ishak Fibrosis Score
from baseline to week 52.
*p = 0.0024
10



























Clinical results at week 104
Overall, clinical results at week 104 in telbivudine-treated patients were consistent with those at
week 52, demonstrating durability of efficacy responses for telbivudine-treated patients with continued
treatment.
Among HBeAg-positive patients, therapeutic response (63% vs 48%; p < 0.0001) and key secondary
endpoints (mean log
10
HBV DNA reduction: -5.74 vs -4.42; p < 0.0001, PCR negativity: 56% vs 39%;
p < 0.0001 and ALT normalisation of 70% vs 62%) demonstrated a widening difference at week 104
between telbivudine and lamivudine, respectively. A trend towards higher rates of HBeAg loss (35%
vs 29%) and seroconversion (30% vs 25%) was also observed for telbivudine. Moreover, in the
subgroup of patients with baseline ALT levels ≥ 2x ULN (320), a significantly higher proportion of
telbivudine patients than lamivudine patients achieved HBeAg seroconversions at week 104 (36% vs
28%, respectively).
Among HBeAg-negative patients, differences in therapeutic response (78% vs 66%) and key
secondary endpoints (mean log
10
HBV DNA reduction: -5.00 vs -4.17, and PCR negativity: 82% vs
57%; p < 0.0001) were higher for telbivudine up to week 104. ALT normalisation rates (78% vs 70%)
continued to be higher by week 104.
Predictability at week 24
At week 24, 203 HBeAg-positive (44%) and 177 HBeAg-negative (80%) telbivudine-treated subjects
achieved non-detectable HBV DNA levels.
For both HBeAg positive and negative patients, week 24 HBV DNA results were a predictor of long-
term favourable outcomes. Telbivudine-treated patients who achieved PCR negativity by week 24 had
the highest rates of PCR negativity and HBeAg seroconversion (in HBeAg-positive patients), and the
lowest overall rates of virological breakthrough at week 104.
Outcome results at week 104, based on level of HBV DNA at week 24, for either HBeAg-positive or
HBeAg-negative patients are presented in Table 7.
Table 7
Key efficacy endpoints at week 104 by serum HBV DNA levels at week 24,
telbivudine patients (NV-02B-007 GLOBE)
HBV DNA at
week 24
Outcome for key efficacy end points at 104 weeks based on week 24 results
Therapeutic
response
n/N (%)
PCR-negative
HBV DNA
n/N (%)
HBeAg
seroconversion
n/N (%)
ALT
normalisation
n/N (%)
Virological
breakthrough*
n/N (%)
HBeAg-positive
< 300 copies/ml
172/203 (85) 166/203 (82) 84/183 (46)
160/194 (82)
22/203 (11)
300 copies/ml to
< 3 log
10
copies/ml
36/57 (63)
35/57 (61)
21/54 (39)
40/54 (74)
18/57 (32)
≥ 3 log
10
copies/ml 82/190 (43)
54/190 (28)
23/188 (12)
106/184 (58)
90/190 (47)
HBeAg-negative
< 300 copies/ml
146/177 (82)
156/177 (88)
N/A
131/159 (82)
11/177 (6)
300 copies/ml to
< 3 log
10
copies/ml
13/18 (72)
14/18 (78)
N/A
13/17 (76)
4/18 (22)
≥ 3 log
10
copies/ml
13/26 (50)
12/26 (46)
N/A
14/26 (54)
12/26 (46)
N/A = not applicable
* Virological breakthrough: “1 log above nadir” definition assessed at week 104
11




















Study NV-02B-015
The efficacy and safety results of the 007 GLOBE study were confirmed in study NV-02B-015. This
study is a phase III, randomised, double-blind study of telbivudine 600 mg once daily compared to
lamivudine 100 mg once daily for a treatment period of 104 weeks in 332 nucleoside-naïve chronic
hepatitis B HBeAg-positive and HBeAg-negative Chinese patients.
Durability of HBeAg-seroconversion
Durability of HBeAg-seroconversion was assessed from pooled data from studies NV-02B-007 and
NV-02B-015. The Kaplan-Meier estimates of the proportion of patients who maintained HBeAg
seroconversion for at least 52 weeks following treatment discontinuation were 86.2% for telbivudine
and 92.8% for lamivudine. Patients included in this analysis had completed ≥ 52 weeks of study drug
treatment and exhibited HBeAg loss for ≥ 24 weeks, with HBV DNA < 5 log
10
copies/ml at the last
visit, or, if they were HBeAg-negative at entry, completed ≥ 52 weeks of study drug treatment AND
had HBsAg loss documented on ≥ 2 consecutive study visits.
Clinical resistance
Analysis of patients with virological rebound (confirmed increase of ≥ 1 log
10
copies/ml HBV DNA
from nadir) in the pivotal study (NV-02B-007) at week 48 indicated that, among HBeAg-positive and
HBeAg-negative patients, 5% (23/458) and 2% (5/222), of telbivudine recipients, respectively, had
virological rebound with detectable HBV resistance mutations. The cumulative rates of genotypically
confirmed telbivudine resistance by week 104 were 25.1% (115/458) for HBeAg-positive patients and
10.8% (24/222) for HBeAg-negative patients.
Out of the 680 telbivudine patients initially included in the pivotal study (NV-02B-007), 517 (76%)
enrolled into study CLDT600A2303 for continued telbivudine treatment for up to 208 weeks. Among
HBeAg-positive patients enrolled into study CLDT600A2303, the 3
rd
and 4
th
year resistance rate was
7.8% (25/321) and 5.9% (19/321), respectively. Similarly in the HBeAg-negative patients, the 3
rd
and
4
th
year resistance rate was 6.1% (12/196) and 4.1% (8/196), respectively.
Table 8
Cumulative virologic breakthrough and genetic resistance by HBeAg status in the
GLOBE study and in study 2303 (year 1 to 4)
GLOBE study patients - ITT
(n = 680)
Study 2303 patients - ITT
(n = 517)
HBeAg
status
Virologic
breakthrough
% (n)
Genotypic
resistance
% (n)
HBeAg
status
Virologic
breakthrough
% (n)
Genotypic
resistance
% (n)
Positive
(n = 458)
41.0 (188)
34.7 (159) Positive
(n = 321)
47.4 (152)
40.8 (131)
Negative
(n = 222)
26.1 (58)
19.8 (44)
Negative
(n = 196)
25.5 (50)
18.9 (37)
ITT = Intent to treat
The GLOBE ITT population included patients who were randomised in the 2-year GLOBE study,
received at least one dose of study treatment and had at least one post-baseline HBV DNA assessment.
The 2303 ITT population included GLOBE study patients who subsequently enrolled into study 2303
for continued telbivudine treatment and had at least one post-baseline HBV DNA assessment in study
2303.
Cumulative calculation:
Two different denominators were used to calculate cumulative rates: 1)
Using the 680 ITT patients in the GLOBE study 2) Using the number of GLOBE patients who
continued into extension study 2303 - 517 ITT patients. The nominators included all patients who
were identified with viral breakthrough/genotypic resistance over 4 years corresponding to each
denominator above.
12












Patients included in study 2303 have detectable (n = 159, HBeAg-postive=135, HBeAg-negative=24)
or undetectable HBV DNA (n = 358). From the 159 patients with detectable HBV DNA, 27 (HBeAg-
positive=23, HBeAg-negative=4) patients developed new resistance mutations during 3
rd
and 4
th
year
of telbivudine treatment.
Genotypic analysis of 203 evaluable sample pairs with HBV DNA ≥ 1,000 copies/ml at week 104
demonstrated that the primary mutation associated with telbivudine resistance was rtM204I, often
associated with mutations rtL180M and rtL80I/V and infrequently with rtV27A, rtL82M, rtV173L,
rtT184I and rtA200V. Baseline factors associated with development of genotypic drug resistance
included: lamivudine treatment, higher baseline HBV DNA, lower baseline serum ALT, and increased
body weight/BMI. On-treatment response parameters at week 24 that predicted emergence of drug
resistant virus by week 104 were HBV DNA > 300 copies/ml and elevation of serum ALT.
Genotypic analysis of 50 HBV isolates from telbivudine-treated patients at week 208 revealed a
similar resistance profile as reported at week 104. Conversions at position 80, 180 and polymorphic
positions 91, 229 were always detected in sequences that harboured the M204I mutation that confers
genotypic resistance. These mutations most likely are compensatory mutations. One isolated rtM204V
mutation and two rtM204I/V/M mutations were reported in telbivudine-treated patients experiencing
viral breakthrough up to week 208. No novel mutation was reported.
When considering patients with viral breakthrough by 104 weeks, the rate of resistance was lower in
patients with HBV DNA < 300 copies/ml at week 24 than in patients with HBV DNA ≥ 300 copies/ml
at week 24. In HBeAg positive patients with HBV DNA < 300 copies/ml at week 24, resistance was
1% (3/203) at 48 weeks and 9% (18/203) at week 104, whilst in patients with HBV DNA
≥ 300 copies/ml resistance was 8% (20/247) at 48 weeks and 39% (97/247) at week 104. In HBeAg-
negative patients with HBV DNA < 300 copies/ml at week 24, resistance was 0% (0/177) at 48 weeks
and 5% (9/177) at week 104, whilst in patients with HBV DNA ≥ 300 copies/ml resistance was 11%
(5/44) at 48 weeks and 34% (15/44) at week 104.
Cross-resistance
Cross-resistance has been observed among HBV nucleoside analogues (see section 4.4). In cell-based
assays, lamivudine-resistant HBV strains containing either the rtM204I mutation or the
rtL180M/rtM204V double mutation had ≥ 1,000-fold reduced susceptibility to telbivudine. HBV
encoding the adefovir resistance-associated substitutions rtN236T or rtA181V had around 0.3- and 4-
fold change in susceptibility to telbivudine in cell culture, respectively (see section 4.4).
5.2 Pharmacokinetic properties
The single- and multiple-dose pharmacokinetics of telbivudine were evaluated in healthy subjects and
in patients with chronic hepatitis B. The pharmacokinetics of telbivudine were not evaluated with the
recommended dose of 600 mg in patients with chronic hepatitis B. However telbivudine
pharmacokinetics are similar between both populations.
Absorption
Following oral administration of a 600 mg single dose of telbivudine to healthy subjects (n = 42), the
peak plasma concentration (C
max
) of telbivudine was 3.2 1.1 g/ml (mean SD) and occurred at
median 3.0 hours post dose. The telbivudine area under the plasma concentration-time curve (AUC
0-∞
)
was 28.0 8.5 g
h/ml (mean SD). Inter-subject variability (CV%) for measures of systemic
exposures (C
max
, AUC) was typically approximately 30%.
Effect of food on oral absorption
Telbivudine absorption and exposure were unaffected when a single 600 mg dose was administered
with food.
Distribution
In vitro
binding of telbivudine to human plasma proteins is low (3.3%).
13
Biotransformation
No metabolites of telbivudine were detected following administration of
14
C-telbivudine in humans.
Telbivudine is not a substrate, inhibitor or inducer of the cytochrome P450 (CYP450) enzyme system.
Elimination
After reaching peak concentration, plasma disposition of telbivudine declined in an bi-exponential
manner with a terminal elimination half-life (t
1/2
) of 41.8 ± 11.8 hours. Telbivudine is eliminated
primarily by urinary excretion of unchanged substance. The renal clearance of telbivudine approaches
normal glomerular filtration rate, suggesting that filtration is the main mechanism of excretion.
Approximately 42% of the dose is recovered in the urine over 7 days following a single 600 mg oral
dose of telbivudine. As renal excretion is the predominant route of elimination, patients with moderate
to severe renal dysfunction and those undergoing haemodialysis require a dose interval adjustment
(see section 4.2).
Linearity / non-linearity
Telbivudine pharmacokinetics are dose proportional over the range of 25 to 1,800 mg. Steady state
was achieved after 5 to 7 days of once-daily administration with an approximate 1.5-fold accumulation
in systemic exposure, suggesting an effective accumulation half-life of approximately 15 hours.
Following once-daily administration of telbivudine 600 mg, steady-state trough plasma concentrations
were approximately 0.2-0.3 g/ml.
Special populations
Gender
There are no significant gender-related differences in telbivudine pharmacokinetics.
Race
There are no significant race-related differences in telbivudine pharmacokinetics.
Paediatrics and geriatrics
Pharmacokinetic studies have not been conducted in paediatric or elderly subjects.
Renal impairment
The single-dose pharmacokinetics of telbivudine (200, 400 and 600 mg) have been evaluated in
patients (without chronic hepatitis B) with various degrees of renal impairment (as assessed by
creatinine clearance). Based on the results shown in Table 9, adjustment of the dose interval for
telbivudine is recommended in patients with creatinine clearance of 50 ml/min (see sections 4.2 and
4.4).
Table 9 Pharmacokinetic parameters (mean
SD) of telbivudine in subjects with various
degrees of renal function
Renal function (creatinine clearance in ml/min)
Normal
(> 80)
(n = 8)
600 mg
Mild (50-80)
(n = 8)
600 mg
Moderate
(30-49)
(n = 8)
400 mg
Severe (< 30)
(n = 6)
200 mg
ESRD/
Haemodialysis
(n = 6)
200 mg
C
max
(g/ml)
3.4 ± 0.9
3.2 ± 0.9
2.8 ± 1.3
1.6 ± 0.8
2.1 ± 0.9
AUC
0-∞
(g•h/ml)
28.5 ± 9.6
32.5 ± 10.1
36.0 ± 13.2
32.5 ± 13.2
67.4 ± 36.9
CL
RENAL
(ml/min)
126.7 ± 48.3
83.3 ± 20.0
43.3 ± 20.0
11.7 ± 6.7
-
14














Renally impaired patients on haemodialysis
Haemodialysis (up to 4 hours) reduces systemic telbivudine exposure by approximately 23%.
Following dose interval adjustment for creatinine clearance, no additional dose modification is
necessary during routine haemodialysis (see section 4.2). Telbivudine should be administered after
haemodialysis.
Hepatic impairment
The pharmacokinetics of telbivudine have been studied in patients (without chronic hepatitis B) with
various degrees of hepatic impairment and in some patients with decompensated liver disease. There
were no significant changes in telbivudine pharmacokinetics in hepatically impaired subjects
compared to unimpaired subjects. Results of these studies indicate that no dosage adjustment is
necessary for patients with hepatic impairment (see section 4.2).
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity and genotoxicity. Telbivudine did not show any carcinogenic
potential. No evidence of a direct toxic effect of telbivudine was seen in standard tests of reproduction
toxicology. In rabbits doses of telbivudine providing exposure levels of 37 times those observed in
humans at the therapeutic dose (600 mg) were associated with an increased incidence of abortion and
early delivery. This effect was considered to be secondary to maternal toxicity.
Fertility was assessed in conventional studies performed in adult rats, and as part of a juvenile
toxicology study.
In adult rats, fertility was reduced when both male and female rats were treated with telbivudine at
doses of 500 or 1000 mg/kg/day (lower fertility index compared to concurrent controls). There were
no abnormalities in sperm morphology or function, and the testes and ovaries were histologically
unremarkable.
No evidence of impaired fertility was seen in other studies when either male or female rats were
treated at doses up to 2000 mg/kg/day and mated with untreated rats (systemic exposure levels
approximately 6-14 times higher than those achieved in humans).
In the juvenile toxicology study, rats were treated from day 14 to day 70 post-partum and were mated
with rats receiving the same treatment (no sibling mating). Fertility was reduced in pairs given
≥ 1000 mg/kg/day as shown by decreases in fertility and mating indices, and reduced conception rate.
However the ovarian and uterine parameters of those females mating successfully were unaffected.
The
no observed adverse effect level
(NOAEL) for effects on fertility or mating parameters amounted
to 250 mg/kg/day, which provided exposure levels 2.5 to 2.8 times higher than those achieved in
humans with normal renal function at the therapeutic dose.
6.
6.1 List of excipients
Tablet core
Cellulose microcrystalline
Povidone
Sodium starch glycolate
Silica, colloidal anhydrous
Magnesium stearate
15
Tablet film coat
Titanium dioxide (E171)
Macrogol
Talc
Hypromellose
6.2 Incompatibilities
Not applicable.
6.3 Shelf life
3 years
6.4 Special precautions for storage
This medicinal product does not require any special storage conditions.
6.5 Nature and contents of container
PVC/aluminium blisters
Pack sizes: 28 or 98 film-coated tablets
Not all pack sizes may be marketed.
6.6 Special precautions for disposal
No special requirements.
7.
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
United Kingdom
8.
EU/1/07/388/001
EU/1/07/388/002
9.
24.04.2007
Detailed information on this product is available on the website of the European Medicines Agency
http://www.ema.europa.eu
16
1.
Sebivo 20 mg/ml oral solution
2.
One ml contains 20 mg telbivudine.
Excipient: A 600 mg dose (30 ml) of oral solution contains approximately 47 mg sodium.
For a full list of excipients, see section 6.1.
3.
Oral solution
Clear, colourless to pale yellow solution.
4.
Sebivo is indicated for the treatment of chronic hepatitis B in adult patients with compensated liver
disease and evidence of viral replication, persistently elevated serum alanine aminotransferase (ALT)
levels and histological evidence of active inflammation and/or fibrosis. See section 5.1 for details of
the study and specific patient characteristics on which this indication is based.
Therapy must be initiated by a physician experienced in the management of chronic hepatitis B
infection.
Adults
The recommended dose of Sebivo is 30 ml, providing a dose equivalent to 600 mg, once daily, taken
orally, with or without food.
Duration of therapy
The optimal treatment duration is unknown. Treatment discontinuation should be considered as
follows:
In HBeAg-positive patients without cirrhosis, treatment should be administered for at least
6-12 months after HBe seroconversion (HBeAg loss and HBV DNA loss with anti-HBe
detection) is confirmed or until HBs seroconversion or there is evidence of loss of efficacy.
Serum ALT and HBV DNA levels should be followed regularly after treatment discontinuation
to detect any late virological relapse.
In HBeAg-negative patients without cirrhosis, treatment should be administered at least until
HBs seroconversion or until there is evidence of loss of efficacy. With prolonged treatment for
more than 2 years, regular reassessment is recommended to confirm that continuation of the
selected therapy remains appropriate for the patient.
On-treatment response at week 24 has been shown to be predictive of longer-term response (see
Table 7 in section 5.1) and might be useful for driving the management of patients treated with
telbivudine monotherapy.
17
Renal impairment
No adjustment of the recommended dose of telbivudine is necessary in patients whose creatinine
clearance is 50 ml/min. Adjustment of the dose is required in patients with creatinine clearance
< 50 ml/min, including those with end-stage renal disease (ESRD) on haemodialysis. A reduction of
the daily dose using Sebivo oral solution, as detailed in Table 1 below, is recommended. If use of the
oral solution is not possible, Sebivo film-coated tablets could be used as an alternative and dosing
should be adjusted by increasing the time interval between doses, as detailed in Table 1.
Table 1
Dosing regimen adjustment of Sebivo in patients with renal impairment
Creatinine clearance
(ml/min)
Telbivudine 20 mg/ml oral
solution
Daily dose adjustment
Telbivudine 600 mg film-coated tablet
Alternative** dose adjustment with
increased dose intervals
50
600 mg (30 ml) once daily
600 mg once daily
30-49
400 mg (20 ml) once daily
600 mg once every 48 hours
< 30 (not requiring
dialysis)
200 mg (10 ml) once daily
600 mg once every 72 hours
ESRD*
120 mg (6 ml) once daily
600 mg once every 96 hours
* End stage renal disease
** In case use of the oral solution is not possible
The proposed dose modifications are based on extrapolation and may not be optimal. The safety and
effectiveness of these dosing adjustment guidelines have not been clinically evaluated. Therefore,
close clinical monitoring is recommended in these patients.
End-stage renal disease patients
For patients with ESRD, Sebivo should be administered after haemodialysis (see section 5.2).
Hepatic impairment
No adjustment to the recommended dose of Sebivo is necessary in patients with hepatic impairment
(see section 5.2).
Children and adolescents
Sebivo is not recommended for use in children and adolescents below 16 years of age due to a lack of
data on safety and efficacy.
Elderly patients (age above 65 years)
No data are available to support a specific dose recommendation for patients over the age of 65 years
(see section 4.4).
Hypersensitivity to the active substance or to any of the excipients.
Combination of telbivudine with pegylated or standard interferon alfa (see sections 4.4 and 4.5).
Severe acute exacerbations of chronic hepatitis B are relatively frequent, and are characterised by
transient elevation of serum ALT. Following initiation of antiviral treatment, serum ALT may rise in
some patients while serum levels of HBV DNA fall (see section 4.8). On average, 4-5 weeks elapsed
prior to the occurrence of an exacerbation in patients treated with telbivudine. Overall, ALT flares
occurred more frequently in HBeAg-positive patients than in HBeAg-negative patients. In patients
with compensated liver disease, this elevation of serum ALT is generally not accompanied by elevated
levels of serum bilirubin or by other signs of hepatic decompensation. The risk of hepatic
18




decompensation – and of a subsequent exacerbation of hepatitis – may be elevated in patients with
cirrhosis. Such patients should therefore be closely monitored.
Exacerbations of hepatitis have also been reported in patients who have terminated treatment of
hepatitis B. Post-treatment ALT flares are normally associated with increases in serum HBV DNA
levels, and the majority of such cases have proven to be self-limiting. Nonetheless, there have also
been reports of severe – and sometimes fatal – post-treatment disease exacerbations. Therefore,
hepatic function should be monitored at regular intervals with both clinical and laboratory follow-up
for at least 6 months after discontinuation of hepatitis B therapy.
Occurrence of lactic acidosis (in the absence of hypoxaemia) sometimes fatal, and usually associated
with severe hepatomegaly with steatosis have been reported with the use of nucleoside/nucleotide
analogues. As telbivudine is a nucleoside analogue, this risk cannot be excluded. Treatment with
nucleoside analogues should be discontinued when rapidly elevating aminotransferase levels,
progressive hepatomegaly or metabolic/lactic acidosis of unknown aetiology occur. Benign digestive
symptoms, such as nausea, vomiting and abdominal pain, may be indicative of lactic acidosis
development. Severe cases, sometimes with fatal outcome, were associated with pancreatitis, liver
failure/hepatic steatosis, renal failure and higher levels of serum lactate. Caution should be exercised
when prescribing nucleoside analogues to any patient (particularly obese women) with hepatomegaly,
hepatitis or other known risk factors for liver disease. These patients should be followed closely.
Muscular effects
Cases of myopathy and myalgia have been reported with telbivudine use several weeks to months after
starting therapy (see section 4.8). Cases of rhabdomyolysis have been reported during post-marketing
use of telbivudine (see section 4.8).
Myopathy, defined as persistent unexplained muscle aches and/or muscle weakness regardless of the
degree of increases in creatine kinase levels, should be considered in any patient with diffuse
unexplained myalgias, muscle tenderness, muscle weakness or myositis (defined as myopathy with
histological evidence of muscle damage). Patients should be advised to report promptly any persistent
unexplained muscle aches, pain, tenderness or weakness. If any of these symptoms are reported, a
detailed muscle examination should be performed in order to evaluate muscle function. Telbivudine
therapy should be discontinued if myopathy is diagnosed.
It is not known whether the risk of myopathy during treatment with telbivudine is increased with
concurrent administration of other medicinal products associated with myopathy (e.g. statins, fibrates,
or ciclosporin). Physicians considering concomitant treatment with other agents associated with
myopathy should weigh carefully the potential benefits and risks and should monitor patients for any
signs or symptoms suggestive of myopathy.
Peripheral neuropathy
Peripheral neuropathy has been uncommonly reported in telbivudine-treated patients. If peripheral
neuropathy is suspected, treatment with telbivudine should be reconsidered (see section 4.8).
An increased risk of developing peripheral neuropathy has been observed in one study when
telbivudine and pegylated interferon alfa-2a were co-administered (see section 4.5). Such increased
risk cannot be excluded for other interferon alfa (pegylated or standard). Moreover, the benefit of the
combination of telbivudine with interferon alfa (pegylated or standard) is not currently established.
Therefore, the combination of telbivudine with pegylated or standard interferon alfa is contraindicated
(see section 4.3).
Renal function
Telbivudine is eliminated primarily by renal excretion, therefore dose interval adjustment is
recommended in patients with creatinine clearance < 50 ml/min, including patients on haemodialysis.
The effectiveness of dosing interval adjustment has not been clinically evaluated. Therefore,
virological response should be closely monitored in patients with increased dosage interval (see
sections 4.2 and 5.2).
19
Patients with cirrhosis without decompensation
Due to the limited data available (about 3% of patients enrolled had cirrhosis), telbivudine should be
used with particular caution in cirrhotic patients. These patients should be closely monitored for
clinical, biochemical and virological parameters associated with hepatitis B during treatment and after
treatment is discontinued.
Patients with cirrhosis with decompensation
There are no efficacy and safety data in patients with decompensated cirrhosis.
Patients with previous exposure to nucleoside/nucleotide analogs
In vitro
, telbivudine was not active against the HBV strains containing rtM204V/rtL180M or rtM204I
mutations (see section 5.1). Telbivudine monotherapy is not an option for patients with established
lamivudine-resistant hepatitis B virus infection. Patients who failed to achieve virological response
following treatment with lamivudine for more than 24 weeks are unlikely to benefit from telbivudine
monotherapy. There is currently no clinical data to properly assess the benefit and risk of switching to
telbivudine for lamivudine-treated patients who achieve complete viral suppression on lamivudine.
There are no data on telbivudine treatment in patients with established adefovir-resistant hepatitis B
virus single mutations of rtN236T or A181V. Results from cell-based assays showed that the adefovir
resistance-associated substitution A181V had 1.5- to approximately 4-fold reduced susceptibility to
telbivudine.
Liver transplant recipients
The safety and efficacy of telbivudine in liver transplant recipients are unknown.
Elderly patients
Clinical studies of telbivudine did not include sufficient numbers of patients 65 years of age to
determine whether they respond differently from younger subjects. In general, caution must be
exercised when prescribing Sebivo to elderly patients in view of the greater frequency of decreased
renal function due to concomitant disease or concomitant use of other medicinal products.
Other special populations
Sebivo has not been investigated in co-infected hepatitis B patients (e.g. patients co-infected with
human immunodeficiency virus [HIV], hepatitis C virus [HCV] or hepatitis D virus [HDV]).
Sebivo oral solution contains approximately 47 mg sodium per 600 mg dose (30 ml), which should be
taken into consideration by patients on a controlled sodium diet.
General
Patients should be advised that treatment with Sebivo has not been shown to reduce the risk of
transmission of HBV to others through sexual contact or blood contamination.
Telbivudine is not recommended to be used with lamivudine because in a phase II study, the treatment
response observed with combination therapy of telbivudine and lamivudine was lower than with
telbivudine alone.
There are currently no efficacy and safety data for other antiviral combinations with telbivudine.
Since telbivudine is eliminated primarily by renal excretion, co-administration of Sebivo with
substances that affect renal function (such as aminoglycosides, loop diuretics, platinum compounds,
vancomycin, amphotericin B) may affect plasma concentrations of telbivudine and/or the co-
administered substance. The combination of telbivudine with these medicinal products should be used
with caution. The steady-state pharmacokinetics of telbivudine were unaltered following multiple dose
administration in combination with lamivudine, adefovir dipivoxil, tenofovir disoproxil fumarate,
20
ciclosporin or pegylated interferon-alfa 2a. In addition, telbivudine does not alter the pharmacokinetics
of lamivudine, adefovir dipivoxil, tenofovir disoproxil fumarate or ciclosporin. No definitive
conclusion could be drawn regarding the effects of telbivudine on the pharmacokinetics of pegylated
interferon due to high interindividual variability of pegylated interferon alfa 2a concentrations. A
clinical trial investigating the combination of telbivudine, 600 mg daily, with pegylated interferon
alfa-2a, 180 micrograms once weekly by subcutaneous administration, indicates that this combination
is associated with an increased risk of developing peripheral neuropathy. The mechanism behind these
events is not known (see section 4.4). The combination of telbivudine with any interferon alfa-
containing product is contraindicated (see section 4.3).
Telbivudine is not a substrate, inhibitor or inducer of the cytochrome P450 (CYP450) enzyme system
(see section 5.2).Therefore, the potential for CYP450-mediated drug interactions involving Sebivo is
low.
For telbivudine no clinical data on exposed pregnancies are available. Animal studies do not indicate
direct harmful effects with respect to pregnancy, embryonal/foetal development, parturition or
postnatal development (see section 5.3). Studies in pregnant rats and rabbits showed that telbivudine
crosses the placenta. Studies in pregnant rabbits showed early delivery and/or abortion secondary to
maternal toxicity. Sebivo should be used during pregnancy only if the benefit to the mother outweighs
the potential risk to the foetus.
There are no data on the effect of telbivudine on transmission of HBV from mother to infant.
Therefore, appropriate interventions should be used to prevent neonatal acquisition of HBV infection.
Telbivudine is excreted in the milk of rats. It is not known whether telbivudine is excreted in human
milk. Women should not breast-feed if they are taking Sebivo.
There are no clinical data on the effects of telbivudine on male or female fertility. In reproductive
toxicology studies in adult animals, fertility was slightly reduced when both male and female rats
received telbivudine. The adverse effects on fertility were greater in a separate study in juvenile
animals when both sexes received telbivudine (see section 5.3).
No studies on the effects on the ability to drive and use machines have been performed.
Assessment of adverse reactions is mainly based on two studies (NV-02B-007 “GLOBE” and NV-
02B-015) in which 1,699 patients with chronic hepatitis B received double-blind treatment with
telbivudine 600 mg/day (n = 847) or lamivudine (n = 852) for 104 weeks.
In the 104-week clinical studies, reported adverse reactions were usually classified as mild or
moderate in severity. The most common adverse events with at least a possible relation to telbivudine
were grade 3/4 blood creatine kinase elevations (6.8%), fatigue (4.4%), headache (3.0%) and nausea
(2.6%).
21
Table 2 lists the adverse reactions according to MedDRA system organ class and frequency using the
following convention: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to
<1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from
the available data). Within each frequency grouping, adverse reactions are presented in order of
decreasing seriousness.
Table 2
Adverse reactions
Metabolism and nutrition disorders
Not known*
Lactic acidosis as a secondary event often
associated with serious conditions (e.g. multi-
organ failure or sepsis)
Nervous system disorders
Common
Dizziness, headache
Uncommon
Peripheral neuropathy, dysgeusia, hypoaesthesia,
paresthesia, sciatica
Respiratory, thoracic and mediastinal
disorders
Common
Cough
Gastrointestinal disorders
Common
Blood amylase increased, diarrhoea, blood lipase
increased, nausea, abdominal pain
Hepatobiliary disorders
Common
Blood alanine aminotransferase increased
Uncommon
Aspartate aminotransferase increased
Skin and subcutaneous tissue disorders
Common
Rash
Musculoskeletal and connective tissue
disorders
Common
Blood creatine phosphokinase increased
Uncommon
Arthralgia, myalgia, myopathy/myositis, pain in
the extremities, back pain, muscle spasm, neck
pain, flank pain
Not known*
Rhabdomyolysis
General disorders and administration site
conditions
Common
Fatigue
Uncommon
Malaise
*
Based on post-marketing reports. Since these reports are from a population of uncertain size it is
not possible to reliably estimate their frequency and therefore the frequency is classed as “not
known”.
Creatine kinase
In the pooled analysis, by 104 weeks of treatment Grade 3/4 creatine kinase elevations (> 7x ULN)
occurred in 12.6% of telbivudine-treated patients and 4.0% of lamivudine-treated patients. Most
creatine kinase elevations were asymptomatic and creatine kinase values typically decreased by the
next visit on continued treatment. In the pivotal study NV-02B-007 (GLOBE), higher pre-treatment
CK values and Caucasian race were identified in both treatment groups as predictive factors for
Grade 3/4 elevations by 104 weeks (see section 4.4).
22
























ALT flares
The incidence of on treatment alanine aminotransferase (ALT) flares in the two treatment arms
according to AASLD (American Association for the Study of Liver Diseases) definition (ALT
elevation > 2x baseline and > 10x ULN) are further described in Table 3 below.
Table 3
Summary of on treatment ALT (IU/L) flare – Pooled NV-02B-007/NV-02B-015
ALT flare:
ALT elevation > 2x baseline and
> 10x ULN
Lamivudine
n/N (%)
Telbivudine
n/N (%)
Overall
67/852 (7.9)
41/847 (4.8)
Baseline to week 24
25/852 (2.9)
25/847 (3.0)
Week 24 to end of study
44/837 (5.3)
17/834 (2.0)
Periodic monitoring of hepatic function is recommended during treatment (see section 4.4).
Exacerbations of hepatitis B after discontinuation of treatment
Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued anti-
hepatitis B therapy including telbivudine (see section 4.4).
The incidence of post-treatment alanine aminotransferase (ALT) flares in the two treatment arms are
further described in Table 4 below.
Table 4
Summary of post-treatment ALT flares – Pooled NV-02B-007/NV-02B-015
Lamivudine
Telbivudine
ALT flare
n/N (%)
n/N (%)
ALT elevation > 2x baseline and > 10x ULN
10/180 (5.6)
9/154 (5.8)
There is no information on intentional overdose of telbivudine, but one subject was given an
unintentional overdose which was asymptomatic. Tested doses up to 1,800 mg/day, three times greater
than the recommended daily dose, have been well tolerated. A maximum tolerated dose of telbivudine
has not been determined. In the event of an overdose, Sebivo should be discontinued and appropriate
general supportive treatment applied as necessary.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Nucleoside and nucleotide reverse transcriptase inhibitors, ATC code:
J05AF11
Telbivudine is a synthetic thymidine nucleoside analogue with activity against HBV DNA
polymerase. It is efficiently phosphorylated by cellular kinases to the active triphosphate form, which
has an intracellular half-life of 14 hours. Telbivudine-5'-triphosphate inhibits HBV DNA polymerase
(reverse transcriptase) by competing with the natural substrate, thymidine 5'-triphosphate.
Incorporation of telbivudine-5'-triphosphate into viral DNA causes DNA chain termination, resulting
in inhibition of HBV replication. Telbivudine is an inhibitor of both HBV first strand
(EC
50
= 0.4-1.3 M) and second strand (EC
50
= 0.12-0.24 M) synthesis, and shows a distinct
preference for inhibiting second strand production. By contrast, telbivudine-5'-triphosphate at
23
















concentrations up to 100 M did not inhibit cellular DNA polymerases , , or . In assays relating to
mitochondrial structure, function and DNA content, telbivudine lacked appreciable toxic effect at
concentrations up to at 10 M and did not increase lactic acid production
in vitro
.
The
in vitro
antiviral activity of telbivudine was assessed in the HBV-expressing human hepatoma cell
line 2.2.15. The concentration of telbivudine that effectively inhibited 50% of viral synthesis (EC
50
)
was approximately 0.2 M. The antiviral activity of telbivudine is specific to the hepatitis B virus and
related hepadnaviruses. Telbivudine was not active against HIV
in vitro
. The absence of activity of
telbivudine against HIV has not been evaluated in clinical trials.
Clinical experience
The safety and efficacy of long-term (104 weeks) Sebivo treatment were evaluated in two active-
controlled clinical studies that included 1,699 patients with chronic hepatitis B (NV-02B-007 GLOBE
and NV-02B-015).
NV-02B-007 GLOBE study
The NV-02B-007 GLOBE study is a randomised, double-blind, multinational phase III study of
telbivudine compared to lamivudine for a treatment period of 104 weeks in 1,367 nucleoside-naïve
chronic hepatitis B HBeAg-positive and HBeAg-negative patients. The majority of the population
enrolled was Asian. The most common HBV genotypes were B (26%) and C (51%). A small number
(total of 98) of Caucasian patients were treated with telbivudine. The primary data analysis was
conducted after all patients had reached week 52.
HBeAg-positive patients
: The mean age of patients was 32 years, 74% were male, 82% were Asian,
12% were Caucasian, and 6% had previously received alfa-interferon therapy.
HBeAg-negative patients
: The mean age of patients was 43 years, 79% were male, 65% were Asian,
23% were Caucasian, and 11% had previously received alfa-interferon therapy.
Clinical results at week 52
Clinical and virological efficacy endpoints were evaluated separately in the HBeAg-positive and
HBeAg-negative patient populations. The primary endpoint of therapeutic response was a composite
serological endpoint requiring suppression of HBV DNA to < 5 log
10
copies/ml in conjunction with
either loss of serum HBeAg or ALT normalised. Secondary endpoints included histological response,
ALT normalisation, and various measures of antiviral efficacy.
Regardless of baseline characteristics, the majority of patients taking Sebivo showed histological,
virological, biochemical, and serological responses to treatment. Baseline ALT levels > 2 x ULN and
baseline HBV DNA < 9 log
10
copies/ml were associated with higher rates of eAg seroconversion in
HBeAg-positive patients. Patients who achieve HBV DNA levels < 3 log
10
copies/ml by week 24 had
optimal responses to treatment; conversely patients with HBV DNA levels > 4 log
10
copies/ml at
24 weeks had less favourable outcomes at week 52.
In HBeAg-positive patients, telbivudine was superior to lamivudine in therapeutic response (75.3% vs
67.0% responders; p = 0.0047). In HBeAg-negative patients, telbivudine was non-inferior to
lamivudine (75.2% and 77.2% responders; p = 0.6187). Caucasian ethnicity was associated with lower
treatment response to both antiviral agents used in the GLOBE trial; however the Caucasian patient
population was very limited (n = 98).
At week 24, 203 HBeAg-positive and 177 HBeAg-negative subjects achieved non-detectable HBV
DNA levels. Of those HBeAg positive subjects, 95% achieved non-detectable HBV DNA, 39%
achieved HBeAg seroconversion, 90% achieved ALT normalisation at week 52 and 0.5% exhibited
resistance at week 48. Similarly of those HBeAg-negative subjects, 96% achieved non-detectable
HBV DNA, 79% achieved ALT normalisation at week 52 and 0% exhibited resistance at week 48.
24
Selected virological, biochemical and serological outcome measures are shown in Table 5 and
histological response in Table 6.
Table 5 Virological, biochemical and serological endpoints at week 52 (NV-02B-007 GLOBE
study)
HBeAg-positive (n = 921)
HBeAg-negative (n = 446)
Response parameter
Telbivudine
600 mg
(n = 458)
Lamivudine
100 mg
(n = 463)
Telbivudine
600 mg
(n = 222)
Lamivudine
100 mg
(n = 224)
Mean HBV DNA
reduction from
baseline (log
10
copies/ml) ± SEM
1,2,3
-6.45 (0.11)
*
-5.54 (0.11)
-5.23 (0.13)
*
-4.40 (0.13)
% Patients HBV
DNA negative by
PCR
60%*
40%
88%*
71%
ALT normalisation
4
77%
75%
74%
79%
HBeAg
seroconversion
4
23%
22%
-
-
HBeAg loss
5
26%
23%
-
-
1
SEM: Standard error of mean
2
Roche COBAS Amplicor
®
PCR Assay (lower limit of quantification 300 copies/ml).
3
HBeAg-positive n = 443 and 444, HBeAg-negative n = 219 and 219, for both telbivudine and
lamivudine groups, respectively. The difference in populations is due to patient discontinuation from
the study and missing HBV DNA assessment at week 52.
4
HBeAg-positive n = 440 and 446, HBeAg-negative n = 203 and 207, for telbivudine and lamivudine
groups, respectively. ALT normalisation assessed only in patients with ALT > ULN at baseline.
5
n = 432 and 442, for telbivudine and lamivudine groups, respectively. HBeAg seroconversion and
loss assessed only in patients with detectable HBeAg at baseline.
*p < 0.0001
Table 6 Histological improvement and change in Ishak Fibrosis Score at week 52 (NV-02B-
007 GLOBE study)
HBeAg-positive (n = 921)
HBeAg-negative (n = 446)
Telbivudine
600 mg
(n = 384)
1
Lamivudine
100 mg
(n = 386)
1
Telbivudine
600 mg
(n = 199)
1
Lamivudine
100 mg
(n = 207)
1
Histological response
2
Improvement
71%*
61%
71%
70%
No improvement
17%
24%
21%
24%
Ishak Fibrosis Score
3
Improvement 42% 47% 49% 45%
No change 39% 32% 34% 43%
Worsening 8% 7% 9% 5%
Missing week 52 biopsy
12% 15% 9% 7%
1
Patients with ≥ one dose of study drug with evaluable baseline liver biopsies and baseline
Knodell Histological Activity Index (HAI) score > 3.
2
Histological response defined as a ≥ 2 point decrease in Knodell Necroinflammatory Score from
baseline with no worsening of the Knodell Fibrosis Score.
3
For Ishak Fibrosis Score, improvement measured as ≥ 1 point reduction in Ishak Fibrosis Score
from baseline to week 52.
*p = 0.0024
25



























Clinical results at week 104
Overall, clinical results at week 104 in telbivudine-treated patients were consistent with those at
week 52, demonstrating durability of efficacy responses for telbivudine-treated patients with continued
treatment.
Among HBeAg-positive patients, therapeutic response (63% vs 48%; p < 0.0001) and key secondary
endpoints (mean log
10
HBV DNA reduction: -5.74 vs -4.42; p < 0.0001, PCR negativity: 56% vs 39%;
p < 0.0001 and ALT normalisation of 70% vs 62%) demonstrated a widening difference at week 104
between telbivudine and lamivudine, respectively. A trend towards higher rates of HBeAg loss (35%
vs 29%) and seroconversion (30% vs 25%) was also observed for telbivudine. Moreover, in the
subgroup of patients with baseline ALT levels ≥ 2x ULN (320), a significantly higher proportion of
telbivudine patients than lamivudine patients achieved HBeAg seroconversions at week 104 (36% vs
28%, respectively).
Among HBeAg-negative patients, differences in therapeutic response (78% vs 66%) and key
secondary endpoints (mean log
10
HBV DNA reduction: -5.00 vs -4.17, and PCR negativity: 82% vs
57%; p < 0.0001) were higher for telbivudine up to week 104. ALT normalisation rates (78% vs 70%)
continued to be higher by week 104.
Predictability at week 24
At week 24, 203 HBeAg-positive (44%) and 177 HBeAg-negative (80%) telbivudine-treated subjects
achieved non-detectable HBV DNA levels.
For both HBeAg positive and negative patients, week 24 HBV DNA results were a predictor of long-
term favourable outcomes. Telbivudine-treated patients who achieved PCR negativity by week 24 had
the highest rates of PCR negativity and HBeAg seroconversion (in HBeAg-positive patients), and the
lowest overall rates of virological breakthrough at week 104.
Outcome results at week 104, based on level of HBV DNA at week 24, for either HBeAg-positive or
HBeAg-negative patients are presented in Table 7.
Table 7
Key efficacy endpoints at week 104 by serum HBV DNA levels at week 24,
telbivudine patients (NV-02B-007 GLOBE)
HBV DNA at
week 24
Outcome for key efficacy end points at 104 weeks based on week 24 results
Therapeutic
response
n/N (%)
PCR-negative
HBV DNA
n/N (%)
HBeAg
seroconversion
n/N (%)
ALT
normalisation
n/N (%)
Virological
breakthrough*
n/N (%)
HBeAg-positive
< 300 copies/ml
172/203 (85) 166/203 (82) 84/183 (46)
160/194 (82)
22/203 (11)
300 copies/ml to
< 3 log
10
copies/ml
36/57 (63)
35/57 (61)
21/54 (39)
40/54 (74)
18/57 (32)
≥ 3 log
10
copies/ml 82/190 (43)
54/190 (28)
23/188 (12)
106/184 (58)
90/190 (47)
HBeAg-negative
< 300 copies/ml
146/177 (82)
156/177 (88)
N/A
131/159 (82)
11/177 (6)
300 copies/ml to
< 3 log
10
copies/ml
13/18 (72)
14/18 (78)
N/A
13/17 (76)
4/18 (22)
≥ 3 log
10
copies/ml
13/26 (50)
12/26 (46)
N/A
14/26 (54)
12/26 (46)
N/A = not applicable
* Virological breakthrough: “1 log above nadir” definition assessed at week 104
26




















Study NV-02B-015
The efficacy and safety results of the 007 GLOBE study were confirmed in study NV-02B-015. This
study is a phase III, randomised, double-blind study of telbivudine 600 mg once daily compared to
lamivudine 100 mg once daily for a treatment period of 104 weeks in 332 nucleoside-naïve chronic
hepatitis B HBeAg-positive and HBeAg-negative Chinese patients.
Durability of HBeAg-seroconversion
Durability of HBeAg-seroconversion was assessed from pooled data from studies NV-02B-007 and
NV-02B-015. The Kaplan-Meier estimates of the proportion of patients who maintained HBeAg
seroconversion for at least 52 weeks following treatment discontinuation were 86.2% for telbivudine
and 92.8% for lamivudine. Patients included in this analysis had completed ≥ 52 weeks of study drug
treatment and exhibited HBeAg loss for ≥ 24 weeks, with HBV DNA < 5 log
10
copies/ml at the last
visit, or, if they were HBeAg-negative at entry, completed ≥ 52 weeks of study drug treatment AND
had HBsAg loss documented on ≥ 2 consecutive study visits.
Clinical resistance
Analysis of patients with virological rebound (confirmed increase of ≥ 1 log
10
copies/ml HBV DNA
from nadir) in the pivotal study (NV-02B-007) at week 48 indicated that, among HBeAg-positive and
HBeAg-negative patients, 5% (23/458) and 2% (5/222), of telbivudine recipients, respectively, had
virological rebound with detectable HBV resistance mutations. The cumulative rates of genotypically
confirmed telbivudine resistance by week 104 were 25.1% (115/458) for HBeAg-positive patients and
10.8% (24/222) for HBeAg-negative patients.
Out of the 680 telbivudine patients initially included in the pivotal study (NV-02B-007), 517 (76%)
enrolled into study CLDT600A2303 for continued telbivudine treatment for up to 208 weeks. Among
HBeAg-positive patients enrolled into study CLDT600A2303, the 3
rd
and 4
th
year resistance rate was
7.8% (25/321) and 5.9% (19/321), respectively. Similarly in the HBeAg-negative patients, the 3
rd
and
4
th
year resistance rate was 6.1% (12/196) and 4.1% (8/196), respectively.
Table 8
Cumulative virologic breakthrough and genetic resistance by HBeAg status in the
GLOBE study and in study 2303 (year 1 to 4)
GLOBE study patients - ITT
(n = 680)
Study 2303 patients - ITT
(n = 517)
HBeAg
status
Virologic
breakthrough
% (n)
Genotypic
resistance
% (n)
HBeAg
status
Virologic
breakthrough
% (n)
Genotypic
resistance
% (n)
Positive
(n = 458)
41.0 (188)
34.7 (159) Positive
(n = 321)
47.4 (152)
40.8 (131)
Negative
(n = 222)
26.1 (58)
19.8 (44)
Negative
(n = 196)
25.5 (50)
18.9 (37)
ITT = Intent to treat
The GLOBE ITT population included patients who were randomised in the 2-year GLOBE study,
received at least one dose of study treatment and had at least one post-baseline HBV DNA assessment.
The 2303 ITT population included GLOBE study patients who subsequently enrolled into study 2303
for continued telbivudine treatment and had at least one post-baseline HBV DNA assessment in study
2303.
Cumulative calculation:
Two different denominators were used to calculate cumulative rates: 1)
Using the 680 ITT patients in the GLOBE study 2) Using the number of GLOBE patients who
continued into extension study 2303 - 517 ITT patients. The nominators included all patients who
were identified with viral breakthrough/genotypic resistance over 4 years corresponding to each
denominator above.
27












Patients included in study 2303 have detectable (n = 159, HBeAg-postive=135, HBeAg-negative=24)
or undetectable HBV DNA (n = 358). From the 159 patients with detectable HBV DNA, 27 (HBeAg-
positive=23, HBeAg-negative=4) patients developed new resistance mutations during 3
rd
and 4
th
year
of telbivudine treatment.
Genotypic analysis of 203 evaluable sample pairs with HBV DNA ≥ 1,000 copies/ml at week 104
demonstrated that the primary mutation associated with telbivudine resistance was rtM204I, often
associated with mutations rtL180M and rtL80I/V and infrequently with rtV27A, rtL82M, rtV173L,
rtT184I and rtA200V. Baseline factors associated with development of genotypic drug resistance
included: lamivudine treatment, higher baseline HBV DNA, lower baseline serum ALT, and increased
body weight/BMI. On-treatment response parameters at week 24 that predicted emergence of drug
resistant virus by week 104 were HBV DNA > 300 copies/ml and elevation of serum ALT.
Genotypic analysis of 50 HBV isolates from telbivudine-treated patients at week 208 revealed a
similar resistance profile as reported at week 104. Conversions at position 80, 180 and polymorphic
positions 91, 229 were always detected in sequences that harboured the M204I mutation that confers
genotypic resistance. These mutations most likely are compensatory mutations. One isolated rtM204V
mutation and two rtM204I/V/M mutations were reported in telbivudine-treated patients experiencing
viral breakthrough up to week 208. No novel mutation was reported.
When considering patients with viral breakthrough by 104 weeks, the rate of resistance was lower in
patients with HBV DNA < 300 copies/ml at week 24 than in patients with HBV DNA ≥ 300 copies/ml
at week 24. In HBeAg positive patients with HBV DNA < 300 copies/ml at week 24, resistance was
1% (3/203) at 48 weeks and 9% (18/203) at week 104, whilst in patients with HBV DNA
≥ 300 copies/ml resistance was 8% (20/247) at 48 weeks and 39% (97/247) at week 104. In HBeAg-
negative patients with HBV DNA < 300 copies/ml at week 24, resistance was 0% (0/177) at 48 weeks
and 5% (9/177) at week 104, whilst in patients with HBV DNA ≥ 300 copies/ml resistance was 11%
(5/44) at 48 weeks and 34% (15/44) at week 104.
Cross-resistance
Cross-resistance has been observed among HBV nucleoside analogues (see section 4.4). In cell-based
assays, lamivudine-resistant HBV strains containing either the rtM204I mutation or the
rtL180M/rtM204V double mutation had ≥ 1,000-fold reduced susceptibility to telbivudine. HBV
encoding the adefovir resistance-associated substitutions rtN236T or rtA181V had around 0.3- and 4-
fold change in susceptibility to telbivudine in cell culture, respectively (see section 4.4).
5.2 Pharmacokinetic properties
The single- and multiple-dose pharmacokinetics of telbivudine were evaluated in healthy subjects and
in patients with chronic hepatitis B. The pharmacokinetics of telbivudine were not evaluated with the
recommended dose of 600 mg in patients with chronic hepatitis B. However telbivudine
pharmacokinetics are similar between both populations.
Absorption
Following oral administration of a 600 mg single dose of telbivudine to healthy subjects (n = 42), the
peak plasma concentration (C
max
) of telbivudine was 3.2 1.1 g/ml (mean SD) and occurred at
median 3.0 hours post dose. The telbivudine area under the plasma concentration-time curve (AUC
0-∞
)
was 28.0 8.5 g
h/ml (mean SD). Inter-subject variability (CV%) for measures of systemic
exposures (C
max
, AUC) was typically approximately 30%. Film-coated tablets containing 600 mg
telbivudine are bioequivalent to 30 ml telbivudine oral solution (20 mg/ml).
Effect of food on oral absorption
Telbivudine absorption and exposure were unaffected when a single 600 mg dose was administered
with food.
28
Distribution
In vitro
binding of telbivudine to human plasma proteins is low (3.3%).
Biotransformation
No metabolites of telbivudine were detected following administration of
14
C-telbivudine in humans.
Telbivudine is not a substrate, inhibitor or inducer of the cytochrome P450 (CYP450) enzyme system.
Elimination
After reaching peak concentration, plasma disposition of telbivudine declined in an bi-exponential
manner with a terminal elimination half-life (t
1/2
) of 41.8 ± 11.8 hours. Telbivudine is eliminated
primarily by urinary excretion of unchanged substance. The renal clearance of telbivudine approaches
normal glomerular filtration rate, suggesting that filtration is the main mechanism of excretion.
Approximately 42% of the dose is recovered in the urine over 7 days following a single 600 mg oral
dose of telbivudine. As renal excretion is the predominant route of elimination, patients with moderate
to severe renal dysfunction and those undergoing haemodialysis require a dose interval adjustment
(see section 4.2).
Linearity / non-linearity
Telbivudine pharmacokinetics are dose proportional over the range of 25 to 1,800 mg. Steady state
was achieved after 5 to 7 days of once-daily administration with an approximate 1.5-fold accumulation
in systemic exposure, suggesting an effective accumulation half-life of approximately 15 hours.
Following once-daily administration of telbivudine 600 mg, steady-state trough plasma concentrations
were approximately 0.2-0.3 g/ml.
Special populations
Gender
There are no significant gender-related differences in telbivudine pharmacokinetics.
Race
There are no significant race-related differences in telbivudine pharmacokinetics.
Paediatrics and geriatrics
Pharmacokinetic studies have not been conducted in paediatric or elderly subjects.
Renal impairment
The single-dose pharmacokinetics of telbivudine (200, 400 and 600 mg) have been evaluated in
patients (without chronic hepatitis B) with various degrees of renal impairment (as assessed by
creatinine clearance). Based on the results shown in Table 9, adjustment of the dose interval for
telbivudine is recommended in patients with creatinine clearance of 50 ml/min (see sections 4.2 and
4.4).
Table 9 Pharmacokinetic parameters (mean
SD) of telbivudine in subjects with various
degrees of renal function
Renal function (creatinine clearance in ml/min)
Normal
(> 80)
(n = 8)
600 mg
Mild (50-80)
(n = 8)
600 mg
Moderate
(30-49)
(n = 8)
400 mg
Severe (< 30)
(n = 6)
200 mg
ESRD/
Haemodialysis
(n = 6)
200 mg
C
max
(g/ml)
3.4 ± 0.9
3.2 ± 0.9
2.8 ± 1.3
1.6 ± 0.8
2.1 ± 0.9
AUC
0-∞
(g•h/ml)
28.5 ± 9.6
32.5 ± 10.1
36.0 ± 13.2
32.5 ± 13.2
67.4 ± 36.9
CL
RENAL
(ml/min)
126.7 ± 48.3
83.3 ± 20.0
43.3 ± 20.0
11.7 ± 6.7
-
29














Renally impaired patients on haemodialysis
Haemodialysis (up to 4 hours) reduces systemic telbivudine exposure by approximately 23%.
Following dose interval adjustment for creatinine clearance, no additional dose modification is
necessary during routine haemodialysis (see section 4.2). Telbivudine should be administered after
haemodialysis.
Hepatic impairment
The pharmacokinetics of telbivudine have been studied in patients (without chronic hepatitis B) with
various degrees of hepatic impairment and in some patients with decompensated liver disease. There
were no significant changes in telbivudine pharmacokinetics in hepatically impaired subjects
compared to unimpaired subjects. Results of these studies indicate that no dosage adjustment is
necessary for patients with hepatic impairment (see section 4.2).
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity and genotoxicity. Telbivudine did not show any carcinogenic
potential. No evidence of a direct toxic effect of telbivudine was seen in standard tests of reproduction
toxicology. In rabbits doses of telbivudine providing exposure levels of 37 times those observed in
humans at the therapeutic dose (600 mg) were associated with an increased incidence of abortion and
early delivery. This effect was considered to be secondary to maternal toxicity.
Fertility was assessed in conventional studies performed in adult rats, and as part of a juvenile
toxicology study.
In adult rats, fertility was reduced when both male and female rats were treated with telbivudine at
doses of 500 or 1000 mg/kg/day (lower fertility index compared to concurrent controls). There were
no abnormalities in sperm morphology or function, and the testes and ovaries were histologically
unremarkable.
No evidence of impaired fertility was seen in other studies when either male or female rats were
treated at doses up to 2000 mg/kg/day and mated with untreated rats (systemic exposure levels
approximately 6-14 times higher than those achieved in humans).
In the juvenile toxicology study, rats were treated from day 14 to day 70 post-partum and were mated
with rats receiving the same treatment (no sibling mating). Fertility was reduced in pairs given
≥ 1000 mg/kg/day as shown by decreases in fertility and mating indices, and reduced conception rate.
However the ovarian and uterine parameters of those females mating successfully were unaffected.
The
no observed adverse effect level
(NOAEL) for effects on fertility or mating parameters amounted
to 250 mg/kg/day, which provided exposure levels 2.5 to 2.8 times higher than those achieved in
humans with normal renal function at the therapeutic dose.
6.
6.1 List of excipients
Benzoic acid
Sodium saccharin
Passion fruit flavouring
Sodium hydroxide
Citric acid anhydrous
Water, purified
30
6.2 Incompatibilities
Not applicable.
6.3 Shelf life
3 years
Use within 2 months of opening the bottle.
6.4 Special precautions for storage
Do not store above 30°C. Do not freeze.
6.5 Nature and contents of container
300 ml brown glass bottle with a child-resistant closure, including a polyethylene sealing disc and a
guarantee ring, a polypropylene dosing cup with embossed graduations from 5 to 30 ml in 5 ml
increments, and a polypropylene oral syringe with graduations of 1 ml to 10 ml in 0.5 ml increments.
6.6 Special precautions for disposal
No special requirements.
7.
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
United Kingdom
8.
EU/1/07/388/003
9.
24.04.2007
Detailed information on this product is available on the website of the European Medicines Agency
http://www.ema.europa.eu
31
ANNEX II
A.
MANUFACTURING AUTHORISATION HOLDER
RESPONSIBLE FOR BATCH RELEASE
B.
CONDITIONS OF THE MARKETING AUTHORISATION
32
A. MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturer responsible for batch release
Novartis Pharma S.A.S.
26, rue de la Chapelle
F-68330 Huningue
France
B. CONDITIONS OF THE MARKETING AUTHORISATION
Medicinal product subject to restricted medical prescription (See Annex I: Summary of Product
Characteristics, section 4.2).
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND
EFFECTIVE USE OF THE MEDICINAL PRODUCT
Not applicable.
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, presented in Module 1.8.1. of the
Marketing Authorisation, is in place and functioning before and whilst the product is on the market.
Risk Management plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in
the Pharmacovigilance Plan, as agreed in version 2 of the Risk Management Plan (RMP) presented in
Module 1.8.2. of the Marketing Authorisation and any subsequent updates of the RMP agreed by the
CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, any
updated RMP should be submitted at the same time as the following Periodic Safety Update Report
(PSUR).
In addition, an updated RMP should be submitted:
When new information is received that may impact on the current Safety Specification,
Pharmacovigilance Plan or risk minimisation activities
Within 60 days of an important (pharmacovigilance or risk minimisation) milestone being
reached
At the request of the European Medicines Agency
33
ANNEX III
LABELLING AND PACKAGE LEAFLET
34
A. LABELLING
35
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
CARTON
1.
Sebivo 600 mg film-coated tablets
telbivudine
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
One tablet contains 600 mg telbivudine.
3.
LIST OF EXCIPIENTS
4.
28 film-coated tablets
98 film-coated tablets
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Oral use.
Read the package leaflet before use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
9.
SPECIAL STORAGE CONDITIONS
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
36

























Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
United Kingdom
EU/1/07/388/001
28 film-coated tablets
EU/1/07/388/002
98 film-coated tablets
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Sebivo 600 mg
37
















MINIMUM PARTICULARS TO APPEAR ON BLISTERS OR STRIPS
BLISTERS
1.
Sebivo 600 mg film-coated tablets
telbivudine
2.
Novartis Europharm Limited
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
OTHER
Monday
Tuesday
Wednesday
Thursday
Friday
Saturday
Sunday
38
















PARTICULARS TO APPEAR ON THE OUTER PACKAGING AND THE IMMEDIATE
PACKAGING
FOLDING BOX AND BOTTLE LABEL
1.
Sebivo 20 mg/ml oral solution
Telbivudine
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each ml contains 20 mg telbivudine.
3.
LIST OF EXCIPIENTS
Also contains sodium. See the package leaflet for further information.
4.
1 bottle containing 300 ml oral solution [folding box only]
1 cup + 1 oral syringe [folding box only]
300 ml [bottle label only]
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Oral use.
Read the package leaflet before use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
Use within 2 months of opening the bottle.
39























9.
SPECIAL STORAGE CONDITIONS
Do not store above 30C.
Do not freeze.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
United Kingdom
EU/1/07/388/003
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Sebivo 20 mg/ml [folding box only]
40




















B. PACKAGE LEAFLET
41
PACKAGE LEAFLET: INFORMATION FOR THE USER
Sebivo 600 mg film-coated tablets
Telbivudine
Read all of this leaflet carefully before you start taking this medicine.
-
If you have any further questions, ask your doctor or pharmacist.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
-
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor or pharmacist.
In this leaflet
:
1.
What Sebivo is and what it is used for
3.
How to take Sebivo
4.
Possible side effects
5.
How to store Sebivo
6.
Further information
1.
WHAT SEBIVO IS AND WHAT IT IS USED FOR
Sebivo belongs to a group of medicines called antiviral medicines, which are used to treat infections
caused by viruses.
Sebivo is used to treat adults with chronic hepatitis B.
Hepatitis B is caused by infection with the hepatitis B virus, which multiplies in the liver and causes
liver damage. Treatment with Sebivo reduces the amount of hepatitis B virus in the body by blocking
its growth, resulting in less liver damage and improved liver function.
2.
BEFORE YOU TAKE SEBIVO
Do not take Sebivo
-
if you are allergic (hypersensitive) to telbivudine or any of the other ingredients of Sebivo
(listed in section 6).
-
if you are being treated with pegylated or standard interferon alfa (see “Taking other
medicines”).
If this applies to you,
do not take Sebivo. Talk to your doctor
. If you think you may be allergic, ask
your doctor for advice.
Take special care with Sebivo
-
if you have or have had any kidney problems. Your doctor may order laboratory tests to check
your kidneys are working properly before and during treatment. Depending on the results of
these tests your doctor may advise you to change how often you take Sebivo.
-
if you suffer from cirrhosis of the liver (a serious condition which causes liver “scarring”). In
this case your doctor will want to monitor you more closely.
-
if you have had a liver transplant.
-
if you are taking any medicines that may cause muscle problems (talk to your doctor or
pharmacist if you are unsure).
-
if you are infected with HIV, hepatitis C or D, or are being treated with any antiviral medicines.
If any of these applies to you,
tell your doctor before you take Sebivo
.
42
-
Keep this leaflet. You may need to read it again.
2.
Before you take Sebivo
-
Sebivo can cause persistent unexplained muscle weakness or muscle pain (myopathy). Muscle
symptoms may progress and become serious, sometimes leading to muscle breakdown
(rhabdomyolysis) which can cause kidney damage.
-
Uncommonly Sebivo can induce numbness, tingling, pain and/or burning sensations in the arms
and/or legs (peripheral neuropathy).
If you experience any of these symptoms during your treatment with Sebivo,
call your doctor
immediately
.
Other effects of this type of medicine
Sebivo belongs to a class of medicines (a nucleoside analogue) that can cause an excess of lactic acid
in the blood (lactic acidosis) and enlargement of the liver (hepatomegaly) with fatty liver (steatosis).
Lactic acidosis is a rare but serious side effect which can occasionally be fatal. Lactic acidosis occurs
more often in women, particularly if they are very overweight. Your doctor will monitor you regularly
while you are receiving Sebivo. If you experience muscle pain, severe and persistent stomach pain
with nausea and vomiting, severe and persistent trouble breathing and tiredness while taking Sebivo,
call your doctor
immediately
.
Some people may get very serious hepatitis symptoms when they stop taking medicines like Sebivo.
Your doctor will monitor your health and do regular blood tests to check your liver after you stop
treatment with Sebivo. Tell your doctor immediately about any new or unusual symptoms that you
notice after stopping treatment (see “If you stop taking Sebivo” in section 3 of this leaflet).
Take care not to infect other people
Sebivo does not reduce the risk of infecting others with hepatitis B virus (HBV) through sexual
contact or exposure to contaminated blood or other body fluids. Never share needles. Do not share
personal items that could have blood or body fluids on them, such as toothbrushes or razor blades. A
vaccine is available to prevent infection with HBV.
Use in children
Sebivo is not recommended for use in children under 16 years of age.
Taking other medicines
Please tell your doctor or pharmacist if you are taking or have recently taken any other medicines,
including medicines obtained without a prescription.
Your doctor or pharmacist needs to know about other medicines because some medicines could affect
your kidneys and because Sebivo mainly leaves the body via the kidneys in the urine.
Do not take Sebivo if you are using pegylated or standard interferon alfa (see “Do not take Sebivo”),
because the combination of these medicines may increase your risk of developing peripheral
neuropathy (numbness, tingling, and/or burning sensations in the arms and/or legs). Tell your doctor
or pharmacist if you are being treated with interferon.
Pregnancy and breast-feeding
-
Do not use Sebivo during pregnancy unless your doctor recommends it. If you are pregnant or
think you may be, tell your doctor before taking Sebivo. Your doctor will discuss with you the
potential risks of taking Sebivo during pregnancy.
-
If you have hepatitis B and become pregnant, talk to your doctor about how you can best protect
your baby. It is not known whether Sebivo reduces the risk of passing your hepatitis B virus on
to your unborn baby.
-
Do not breast-feed during treatment with Sebivo. Tell your doctor if you are breast-feeding.
43
3.
HOW TO TAKE SEBIVO
Always take Sebivo exactly as your doctor has told you. You should check with your doctor or
pharmacist if you are not sure.
How much Sebivo to take
The usual dose of Sebivo is one 600 mg tablet once a day. Take the tablet at about the same time each
day.
The tablet can be taken with or without food. Swallow it whole with some water. Do not chew, split or
crush it.
You may need to take Sebivo less frequently if you have kidney problems. Tell your doctor if you
have, or have ever had, any kidney problems.
How long to take Sebivo
Do not change your dose or stop taking Sebivo without talking to your doctor. Take Sebivo every day
and continue your treatment exactly as prescribed unless your doctor says otherwise. Your doctor will
make regular checks to make sure that Sebivo is working. Your hepatitis B symptoms may get worse
or become very serious if you stop taking Sebivo.
If you take more Sebivo than you should
If you have taken too much Sebivo, or if someone else accidentally takes your tablets, go to your
doctor or hospital for advice straight away. Take the pack of tablets with you and show it to your
doctor.
If you forget to take Sebivo
-
If you forget to take Sebivo, take it as soon as you remember and then take your next dose at its
regular time.
-
However, if it is almost time for your next dose, skip the dose you missed and take the next one
at the usual time.
Do not take a double dose to make up for a forgotten tablet. This may increase the chance of you
getting unwanted side effects. Ask your doctor or pharmacist if you are not sure what to do.
If you stop taking Sebivo
Stopping treatment with Sebivo may result in a worsening of your hepatitis B infection. Do not stop
Sebivo unless your doctor tells you to. While you are taking Sebivo, make sure you do not run out of
Sebivo.
Your doctor will monitor your health and do regular blood tests to check your liver after you stop
treatment with Sebivo since your hepatitis B infection may get worse or become very serious after
stopping treatment. Tell your doctor immediately about any new or unusual symptoms that you notice
after stopping treatment.
4.
POSSIBLE SIDE EFFECTS
Like all medicines, Sebivo can cause side effects, although not everybody gets them.
The frequencies are defined as:
very common:
affects more than 1 patient in 10
uncommon:
affects 1 to 10 patients in 1,000
rare:
affects 1 to 10 patients in 10,000
very rare:
affects less than 1 patient in 10,000
not known:
frequency cannot be estimated from the available data.
44
common:
affects 1 to 10 patients in 100
Some uncommon side effects could be serious:
-
Persistent muscle weakness or muscle pain
-
Numbness, tingling, pain and/or burning sensation in the arms and/or legs
If you experience any of these,
call your doctor immediately
.
Sebivo may also cause other side effects:
Common side effects
-
Dizziness, headache
-
Cough
-
Diarrhoea, feeling sick (nausea), stomach (abdominal) pain
-
Skin rash
-
Tiredness (fatigue)
-
Blood test results show higher levels of liver enzymes, amylase, lipase or creatine kinase
Uncommon side effects
-
Joint pain
-
Persistent muscle weakness or muscle pain (myopathy/myositis), muscle cramp
-
Back, neck and flank pain
-
Numbness, tingling, pain and/or burning sensation in the arms and/or legs or around the mouth
-
Sciatica (pain in lower back or hip and may radiate into the leg)
-
Taste disturbance
-
Feeling unwell (malaise)
Frequency not known
Excess of lactic acid in the blood (lactic acidosis) and muscle breakdown (rhabdomyolysis) have been
reported.
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet, please
tell your doctor or pharmacist.
5.
HOW TO STORE SEBIVO
Keep out of the reach and sight of children.
Do not use Sebivo after the expiry date which is stated on the carton after EXP. The expiry date refers
to the last day of that month.
Do not use if the pack is damaged or shows signs of tampering.
Medicines should not be disposed of via wastewater or household waste. Ask your pharmacist how to
dispose of medicines no longer required. These measures will help to protect the environment.
6.
FURTHER INFORMATION
What Sebivo contains
-
The other ingredients are: cellulose, microcrystalline; povidone; sodium starch glycolate; silica,
colloidal anhydrous; magnesium stearate; hypromellose; titanium dioxide (E171); talc;
macrogol.
What Sebivo looks like and contents of the pack
Sebivo film-coated tablets are white to slightly yellowish, oval, film-coated tablets with “LDT”
imprinted on one side.
45
-
The active substance is telbivudine. Each tablet contains 600 mg telbivudine.
Sebivo film-coated tablets are supplied in packs of 28 or 98 tablets. Not all pack sizes may be
marketed in your country.
Marketing Authorisation Holder
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
United Kingdom
Manufacturer
Novartis Pharma S.A.S.
26, rue de la Chapelle
F-68330 Huningue
France
For any information about this medicine, please contact the local representative of the Marketing
Authorisation Holder:
België/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Luxembourg/Luxemburg
Novartis Pharma GmbH
Tel: +49 911 273 0
България
Novartis Pharma Services Inc.
Тел.: +359 2 489 98 28
Magyarország
Novartis Hungária Kft. Pharma
Tel.: +36 1 457 65 00
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Malta
Novartis Pharma Services Inc.
Tel: +356 2298 3217
Danmark
Novartis Healthcare A/S
Tlf: +45 39 16 84 00
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Deutschland
Novartis Pharma GmbH
Tel: +49 911 273 0
Norge
Novartis Norge AS
Tlf: +47 23 05 20 00
Eesti
Novartis Pharma Services Inc.
Tel: +372 66 30 810
Österreich
Novartis Pharma GmbH
Tel: +43 1 86 6570
Ελλά
Novartis (Hellas) A.E.B.E.
Τηλ: +30 210 281 17 12
Polska
Novartis Poland Sp. z o.o.
Tel.: +48 22 550 8888
España
Novartis Farmacéutica, S.A.
Tel: +34 93 306 42 00
Portugal
Novartis Farma - Produtos Farmacêuticos, S.A.
Tel: +351 21 000 8600
France
Novartis Pharma S.A.S.
Tél: +33 1 55 47 66 00
România
Novartis Pharma Services Inc.
Tel: +40 21 31299 01
46
Ireland
Novartis Ireland Limited
Tel: +353 1 260 12 55
Slovenija
Novartis Pharma Services Inc.
Tel: +386 1 300 75 50
Ísland
Vistor hf.
Sími: +354 535 7000
Slovenská republika
Novartis Slovakia s.r.o.
Tel: +421 2 5542 5439
Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Suomi/Finland
Novartis Finland Oy
Puh/Tel: +358 (0)10 6133 200
Κύπρς
Novartis Pharma Services Inc.
Τηλ: +357 22 690 690
Sverige
Novartis Sverige AB
Tel: +46 8 732 32 00
Latvija
Novartis Pharma Services Inc.
Tel: +371 67 887 070
United Kingdom
Novartis Pharmaceuticals UK Ltd.
Tel: +44 1276 698370
Lietuva
Novartis Pharma Services Inc.
Tel: +370 5 269 16 50
This leaflet was last approved in
Detailed information on this medicine is available on the European Medicines Agency website:
http://www.ema.europa.eu
47
PACKAGE LEAFLET: INFORMATION FOR THE USER
Sebivo 20 mg/ml oral solution
Telbivudine
Read all of this leaflet carefully before you start taking this medicine.
-
If you have any further questions, ask your doctor or pharmacist.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
-
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor or pharmacist.
In this leaflet
:
1.
What Sebivo is and what it is used for
3.
How to take Sebivo
4.
Possible side effects
5.
How to store Sebivo
6.
Further information
1.
WHAT SEBIVO IS AND WHAT IT IS USED FOR
Sebivo belongs to a group of medicines called antiviral medicines, which are used to treat infections
caused by viruses.
Sebivo is used to treat adults with chronic hepatitis B.
Hepatitis B is caused by infection with the hepatitis B virus, which multiplies in the liver and causes
liver damage. Treatment with Sebivo reduces the amount of hepatitis B virus in the body by blocking
its growth, resulting in less liver damage and improved liver function.
2.
BEFORE YOU TAKE SEBIVO
Do not take Sebivo
-
if you are allergic (hypersensitive) to telbivudine or any of the other ingredients of Sebivo
(listed in section 6).
-
if you are being treated with pegylated or standard interferon alfa (see “Taking other
medicines”).
If this applies to you,
do not take Sebivo. Talk to your doctor
. If you think you may be allergic, ask
your doctor for advice.
Take special care with Sebivo
-
if you have or have had any kidney problems. Your doctor may order laboratory tests to check
your kidneys are working properly before and during treatment. Depending on the results of
these tests your doctor may advise you to change how often you take Sebivo.
-
if you suffer from cirrhosis of the liver (a serious condition which causes liver “scarring”). In
this case your doctor will want to monitor you more closely.
-
if you have had a liver transplant.
-
if you are taking any medicines that may cause muscle problems (talk to your doctor or
pharmacist if you are unsure).
-
if you are infected with HIV, hepatitis C or D, or are being treated with any antiviral medicines.
If any of these applies to you,
tell your doctor before you take Sebivo.
48
-
Keep this leaflet. You may need to read it again.
2.
Before you take Sebivo
-
Sebivo can cause persistent unexplained muscle weakness or muscle pain (myopathy). Muscle
symptoms may progress and become serious, sometimes leading to muscle breakdown
(rhabdomyolysis) which can cause kidney damage.
-
Uncommonly Sebivo can cause numbness, tingling, pain and/or burning sensations in the arms
and/or legs (peripheral neuropathy).
If you experience any of these symptoms during your treatment with Sebivo,
call your doctor
immediately
.
Other effects of this type of medicine
Sebivo belongs to a class of medicines (a nucleoside analogue) that can cause an excess of lactic acid
in the blood (lactic acidosis) and enlargement of the liver (hepatomegaly) with fatty liver (steatosis).
Lactic acidosis is a rare but serious side effect which can occasionally be fatal. Lactic acidosis occurs
more often in women, particularly if they are very overweight. Your doctor will monitor you regularly
while you are receiving Sebivo. If you experience muscle pain, severe and persistent stomach pain
with nausea and vomiting, severe and persistent trouble breathing and tiredness while taking Sebivo,
call your doctor immediately
.
Some people may get very serious hepatitis symptoms when they stop taking medicines like Sebivo.
Your doctor will monitor your health and do regular blood tests to check your liver after you stop
treatment with Sebivo. Tell your doctor immediately about any new or unusual symptoms that you
notice after stopping treatment (see “If you stop taking Sebivo” in section 3 of this leaflet).
Take care not to infect other people
Sebivo does not reduce the risk of infecting others with hepatitis B virus (HBV) through sexual
contact or exposure to contaminated blood or other body fluids. Never share needles. Do not share
personal items that could have blood or body fluids on them, such as toothbrushes or razor blades. A
vaccine is available to prevent infection with HBV.
Use in children
Sebivo is not recommended for use in children under 16 years of age.
Taking other medicines
Please tell your doctor or pharmacist if you are taking or have recently taken any other medicines,
including medicines obtained without a prescription.
Your doctor or pharmacist needs to know about other medicines because some medicines could affect
your kidneys and because Sebivo mainly leaves the body via the kidneys in the urine.
Do not take Sebivo if you are using pegylated or standard interferon alfa (see “Do not take Sebivo”),
because the combination of these medicines may increase your risk of developing peripheral
neuropathy (numbness, tingling, and/or burning sensations in the arms and/or legs). Tell your doctor
or pharmacist if you are being treated with interferon.
Taking Sebivo with food and drink
You can take Sebivo with or without food.
Pregnancy and breast-feeding
-
Do not use Sebivo during pregnancy unless your doctor recommends it. If you are pregnant or
think you may be, tell your doctor before taking Sebivo. Your doctor will discuss with you the
potential risks of taking Sebivo during pregnancy.
-
If you have hepatitis B and become pregnant, talk to your doctor about how you can best protect
your baby. It is not known whether Sebivo reduces the risk of passing your hepatitis B virus on
to your unborn baby.
-
Do not breast-feed during treatment with Sebivo. Tell your doctor if you are breast-feeding.
49
Important information about some of the ingredients of Sebivo
Sebivo oral solution contains approximately 47 mg of sodium per 600 mg dose (30 ml). If you are on a
controlled sodium diet, ask your doctor for advice.
3.
HOW TO TAKE SEBIVO
Always take Sebivo exactly as your doctor has told you. You should check with your doctor or
pharmacist if you are not sure.
How much Sebivo to take
The usual dose of Sebivo is 30 ml of oral solution (600 mg telbivudine) once a day. Take Sebivo at
about the same time each day. It can be taken with or without food.
For full instructions on how to take Sebivo, see section “Instructions for use” at the end of this leaflet.
Remove the dosing cup and open the bottle. Slowly and carefully pour the solution from the bottle into
the dosing cup until it reaches the prescribed amount. Swallow the entire contents of the dosing cup
immediately.
If you cannot measure the prescribed amount precisely using the dosing cup alone, you should use the
oral syringe. Detailed instructions on how to use this are given in the section “Instructions for use”.
Your dose may be reduced if you have kidney problems. Tell your doctor if you have, or have ever
had, any kidney problems.
How long to take Sebivo
Do not change your dose or stop taking Sebivo without talking to your doctor. Take Sebivo every day
and continue your treatment exactly as prescribed unless your doctor says otherwise. Your doctor will
make regular checks to make sure that Sebivo is working. Your hepatitis B symptoms may get worse
or become very serious if you stop taking Sebivo.
If you take more Sebivo than you should
If you have taken too much Sebivo, or if someone else accidentally takes your oral solution, go to your
doctor or hospital for advice straight away. Take the pack with you and show it to the doctor.
If you forget to take Sebivo
-
If you forget to take Sebivo, take it as soon as you remember and then take your next dose at its
regular time.
-
However, if it is almost time for your next dose, skip the dose you missed and take the next one
at the usual time.
Do not take a double dose to make up for a forgotten dose. This may increase the chance of you
getting unwanted side effects. Ask your doctor or pharmacist if you are not sure what to do.
If you stop taking Sebivo
Stopping treatment with Sebivo may result in a worsening of your hepatitis B infection. Do not stop
Sebivo unless your doctor tells you to. While you are taking Sebivo, make sure you do not run out of
Sebivo.
Your doctor will monitor your health and do regular blood tests to check your liver after you stop
treatment with Sebivo since your hepatitis B infection may get worse or become very serious after
stopping treatment. Tell your doctor immediately about any new or unusual symptoms that you notice
after stopping treatment.
50
4.
POSSIBLE SIDE EFFECTS
Like all medicines, Sebivo can cause side effects, although not everybody gets them.
The frequencies are defined as:
very common:
affects more than 1 patient in 10
common:
affects 1 to 10 patients in 100
uncommon:
affects 1 to 10 patients in 1,000
very rare:
affects less than 1 patient in 10,000
not known:
frequency cannot be estimated from the available data.
Some uncommon side effects could be serious:
-
Persistent muscle weakness or muscle pain
-
Numbness, tingling, pain and/or burning sensation in the arms and/or legs
If you experience any of these,
call your doctor immediately
.
Sebivo may also cause other side effects:
Common side effects
-
Dizziness, headache
-
Cough
-
Diarrhoea, feeling sick (nausea), stomach (abdominal) pain
-
Tiredness (fatigue)
-
Blood test results show higher levels of liver enzymes, amylase, lipase or creatine kinase
Uncommon side effects
-
Joint pain
-
Persistent muscle weakness or muscle pain (myopathy/myositis), muscle cramp
-
Numbness, tingling, pain and/or burning sensation in the arms and/or legs or around the mouth
-
Sciatica (pain in lower back or hip and may radiate into the leg)
-
Taste disturbance
-
Feeling unwell (malaise)
Frequency not known
Excess of lactic acid in the blood (lactic acidosis) and muscle breakdown (rhabdomyolysis) have been
reported.
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet, please
tell your doctor or pharmacist.
5.
HOW TO STORE SEBIVO
Keep out of the reach and sight of children.
Do not use Sebivo after the expiry date which is stated on the carton and bottle label. The expiry date
refers to the last day of that month.
Do not store above 30C. Do not freeze.
Use within 2 months of opening the bottle.
Ask your pharmacist how to dispose of medicines no longer required. These measures will help to
protect the environment.
51
rare:
affects 1 to 10 patients in 10,000
-
Skin rash
-
Back, neck and flank pain
6.
FURTHER INFORMATION
What Sebivo contains
-
The active substance is telbivudine. 30 ml oral solution contain 600 mg telbivudine.
-
The other ingredients are: benzoic acid, sodium saccharin, passion fruit flavouring, sodium
hydroxide, citrc acid anhydrous, water.
What Sebivo looks like and contents of the pack
Sebivo 20 mg/ml oral solution is supplied as 300 ml of a clear, colourless to pale yellow solution in an
brown glass bottle with a child-resistant white polypropylene closure, including a polyethylene sealing
disc and a guarantee ring. The pack contains an oral dosing cup made of polypropylene with embossed
graduations from 5 to 30 ml in 5 ml increments and a polypropylene oral syringe with graduations of
1 ml to 10 ml in 0.5 ml increments.
Marketing Authorisation Holder
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
United Kingdom
Manufacturer
Novartis Pharma S.A.S.
26, rue de la Chapelle
F-68330 Huningue
France
For any information about this medicine, please contact the local representative of the Marketing
Authorisation Holder:
België/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Luxembourg/Luxemburg
Novartis Pharma GmbH
Tel: +49 911 273 0
България
Novartis Pharma Services Inc.
Тел.: +359 2 489 98 28
Magyarország
Novartis Hungária Kft. Pharma
Tel.: +36 1 457 65 00
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Malta
Novartis Pharma Services Inc.
Tel: +356 2298 3217
Danmark
Novartis Healthcare A/S
Tlf: +45 39 16 84 00
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Deutschland
Novartis Pharma GmbH
Tel: +49 911 273 0
Norge
Novartis Norge AS
Tlf: +47 23 05 20 00
Eesti
Novartis Pharma Services Inc.
Tel: +372 66 30 810
Österreich
Novartis Pharma GmbH
Tel: +43 1 86 6570
52
Ελλά
Novartis (Hellas) A.E.B.E.
Τηλ: +30 210 281 17 12
Polska
Novartis Poland Sp. z o.o.
Tel.: +48 22 550 8888
España
Novartis Farmacéutica, S.A.
Tel: +34 93 306 42 00
Portugal
Novartis Farma - Produtos Farmacêuticos, S.A.
Tel: +351 21 000 8600
France
Novartis Pharma S.A.S.
Tél: +33 1 55 47 66 00
România
Novartis Pharma Services Inc.
Tel: +40 21 31299 01
Ireland
Novartis Ireland Limited
Tel: +353 1 260 12 55
Slovenija
Novartis Pharma Services Inc.
Tel: +386 1 300 75 50
Ísland
Vistor hf.
Sími: +354 535 7000
Slovenská republika
Novartis Slovakia s.r.o.
Tel: +421 2 5542 5439
Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Suomi/Finland
Novartis Finland Oy
Puh/Tel: +358 (0)10 6133 200
Κύπρς
Novartis Pharma Services Inc.
Τηλ: +357 22 690 690
Sverige
Novartis Sverige AB
Tel: +46 8 732 32 00
Latvija
Novartis Pharma Services Inc.
Tel: +371 67 887 070
United Kingdom
Novartis Pharmaceuticals UK Ltd.
Tel: +44 1276 698370
Lietuva
Novartis Pharma Services Inc.
Tel: +370 5 269 16 50
This leaflet was last approved in
Detailed information on this medicine is available on the European Medicines Agency website:
http://www.ema.europa.eu
53
INSTRUCTIONS FOR USE
Please read these instructions carefully so that you know how to use the solution correctly.
1. Bottle containing the oral solution.
2. Child-resistant screw cap with a guarantee-ring.
Always close the bottle with the cap after use.
3. Oral dosing cup for measuring out the dose.
Always put the dosing cup back onto the cap
after use and cleaning.
4. Oral syringe for measuring out doses that cannot
be precisely measured using the cup.
Preparing a dose of medicine using the dosing cup
1.
Remove the dosing cup.
2.
Simultaneously press down (2a) and turn the child-resistant
cap (2b) to open the bottle.
3.
Before pouring any solution into the cup, please check the
position of the appropriate graduation to avoid any potential
waste or spillage.
Holding the cup at eye level, carefully and slowly pour the
prescribed amount of solution from the bottle into the dosing
cup until the solution reaches the top of the appropriate
graduation.
Note
: If the amount poured into the cup exceeds the required dose,
discard the excess in the sink. Do not pour it back into the bottle.
4.
Drink the solution or administer it to the patient immediately.
5.
Close the bottle by screwing the cap back on tightly.
54


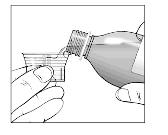
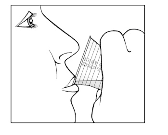
6.
Immediately rinse the dosing cup with water.
7.
Remove the water from the dosing cup,
wipe it with a clean
tissue and place it back on top of the cap.
Preparing a 6 ml dose of medicine using the oral syringe
1.
Remove the dosing cup.
2.
Simultaneously press down (2a) and turn the child-
resistant cap (2b) to open the bottle.
3.
Before pouring any solution into the cup, please
check the position of the 5 and 10 ml marks to avoid
any potential waste or spillage.
Holding the cup at eye level, carefully and slowly
pour the solution from the bottle into the cup until it
comes to about halfway between the 5 ml and 10 ml
marks.
4.
Withdraw all the solution from the cup into the
syringe.
5.
Turn the syringe to the upright position and incline it
slightly so that the air bubbles rise to the top.
6.
Push the plunger carefully and slowly to remove the
air until a droplet of solution appears.
55



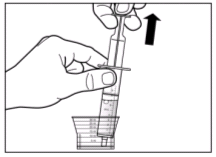

7.
Hold the syringe above the cup.
8.
Push the plunger slowly and carefully until the
solution reaches the 6 ml mark.
9.
Immediately swallow the solution direct from the
syringe.
10.
Discard the solution left in the cup into the sink. Do
not pour it back into the bottle as this could cause
contamination.
11.
Close the bottle firmly.
12.
Rinse the cup and syringe with clean water.
13.
Dry the cup with a clean tissue and place it back over
the cap of the bottle.
14.
Allow the syringe to air-dry and store with the bottle.
56


Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/sebivo.html
Copyright © 1995-2021 ITA all rights reserved.