










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Vectibix 20 mg/ml concentrate for solution for infusion.
2.
Each ml of concentrate contains 20 mg panitumumab.
Each vial contains either 100 mg of panitumumab in 5 ml, 200 mg in 10 ml, or 400 mg in 20 ml.
When prepared according to the instructions given in section 6.6, the final panitumumab concentration
should not exceed 10 mg/ml.
Panitumumab is a human monoclonal IgG2 antibody produced in a mammalian cell line (CHO) by
recombinant DNA technology.
Excipient:
Each ml of concentrate contains 0.150 mmol sodium, which is 3.45 mg sodium.
For a full list of excipients, see section 6.1.
3.
Concentrate for solution for infusion.
Colourless solution that may contain, translucent to white, visible amorphous, proteinaceous
panitumumab particles.
Vectibix is indicated as monotherapy for the treatment of patients with EGFR expressing metastatic
colorectal carcinoma with non-mutated (wild-type)
KRAS
after failure of fluoropyrimidine-,
oxaliplatin-, and irinotecan-containing chemotherapy regimens.
Posology
Vectibix treatment should be supervised by a physician experienced in the use of anti-cancer therapy.
Detection of non-mutated
KRAS
expression should be performed by an experienced laboratory using a
validated test method.
The recommended dose of Vectibix is 6 mg/kg of bodyweight given once every two weeks. Prior to
infusion, Vectibix should be diluted in 0.9% sodium chloride injection to a final concentration not to
exceed 10 mg/ml (for preparation instructions see section 6.6).
Method of administration
Vectibix must be administered as an intravenous (IV) infusion via an infusion pump, using a low
protein binding 0.2 or 0.22 micrometer in-line filter, through a peripheral line or indwelling catheter.
The recommended infusion time is approximately 60 minutes. Doses higher than 1000 mg should be
infused over approximately 90 minutes (for handling instructions, see section 6.6).
2
The infusion line should be flushed with sodium chloride solution before and after Vectibix
administration to avoid mixing with other medicinal products or IV solutions.
Do not administer as an IV push or bolus.
For instructions on dilution of the medicinal product before administration, see section 6.6.
Special populations
The safety and efficacy of Vectibix have not been studied in patients with renal or hepatic impairment.
Dose adjustment is not required in the elderly. In clinical studies no overall differences in safety or
efficacy were observed between patients aged ≥ 65 years and younger patients.
There is no experience in children and Vectibix should not be used in those patients less than 18 years
of age.
Vectibix is contraindicated in patients with a history of severe or life-threatening hypersensitivity
reactions to the active substance or to any of the excipients (see section 4.4).
Patients with interstitial pneumonitis or pulmonary fibrosis (see section 4.4).
Dermatological reactions
Dermatologic related reactions, a pharmacologic effect observed with epidermal growth factor
receptor (EGFR) inhibitors, are experienced with nearly all patients (approximately 90%) treated with
Vectibix (see section 4.8), the majority are mild to moderate in nature. If a patient develops
dermatologic reactions that are grade 3 (NCI-CTC/CTCAE) or higher, or that are considered
intolerable, temporarily withhold Vectibix administration until the reactions have improved
(≤ grade 2). Once improved to ≤ grade 2, reinstate Vectibix administration at 50% of the original dose.
If reactions do not recur, escalate the dose of Vectibix by 25% increments until the recommended dose
is reached. If reactions do not resolve (to ≤ grade 2) after withholding 1 or 2 doses of Vectibix, or if
reactions recur or become intolerable at 50% of the original dose, the use of Vectibix should be
permanently discontinued.
In clinical studies, subsequent to the development of severe dermatological reactions (including
stomatitis), infectious complications including sepsis, in rare cases leading to death, and local
abscesses requiring incisions and drainage were reported. Patients who have severe dermatologic
reactions or who develop worsening reactions whilst receiving Vectibix should be monitored for the
development of inflammatory or infectious sequelae (including cellulitis), and appropriate treatment
promptly initiated. It is recommended that patients wear sunscreen and hats and limit sun exposure
whilst receiving Vectibix and experiencing rash/dermatological toxicities, as sunlight can exacerbate
any skin reactions that may occur.
Pulmonary complications
Patients with a history of, or evidence of, interstitial pneumonitis or pulmonary fibrosis were excluded
from clinical studies. As Interstitial Lung Disease (ILD) has been observed with EGFR inhibitors, in
the event of acute onset or worsening pulmonary symptoms, Vectibix treatment should be interrupted
and a prompt investigation of these symptoms should occur. If pneumonitis or lung infiltrates are
diagnosed, Vectibix should be discontinued and the patient should be treated appropriately.
3
Electrolyte disturbances
Progressively decreasing serum magnesium levels leading to severe (grade 4) hypomagnesaemia have
been observed in some patients. Patients should be periodically monitored for hypomagnesaemia and
accompanying hypocalcaemia prior to initiating Vectibix treatment, and periodically thereafter for up
to 8 weeks after the completion of treatment (see section 4.8). Magnesium repletion is recommended,
as appropriate.
Other electrolyte disturbances, including hypokalaemia, have also been observed. Repletion of these
electrolytes is also recommended, as appropriate.
Infusion related reactions
In a clinical study, 4% of patients experienced infusion-related reactions, and in 1% of patients, these
reactions were graded as severe (NCI-CTC grade 3 and 4).
Across all clinical studies, infusion-related reactions (occurring within 24 hours of any infusion), were
reported in 3% of Vectibix-treated patients, of which < 1% were severe (NCI-CTC grade 3 and 4). In
the post-marketing setting, serious infusion-related reactions have been reported, including rare post-
marketing reports with a fatal outcome. If a severe or life-threatening reaction occurs during an
infusion or at any time post-infusion [eg., presence of bronchospasm, angioedema, hypotension, need
for parenteral medication, or anaphylaxis], Vectibix should be permanently discontinued (see sections
4.3 and 4.8).
In patients experiencing a mild or moderate (NCI-CTC grade 1 and 2) infusion-related reaction the
infusion rate should be reduced for the duration of that infusion. It is recommended to maintain this
lower infusion rate in all subsequent infusions.
Hypersensitivity reactions occurring more than 24 hours after infusion have been reported including a
fatal case of angioedema that occurred more than 24 hours after the infusion. Patients should be
informed of the possibility of a late onset reaction and instructed to contact their physician if
symptoms of a hypersensitivity reaction occur.
Other precautions
This medicinal product contains 0.150 mmol sodium (which is 3.45 mg sodium) per ml of concentrate.
To be taken into consideration by patients on a controlled sodium diet.
Vectibix in combination with IFL
Patients receiving Vectibix in combination with the IFL regimen [bolus 5-fluorouracil (500 mg/m
2
),
leucovorin (20 mg/m
2
) and irinotecan (125 mg/m
2
)] experienced a high incidence of severe diarrhoea
(see section 4.8). Therefore administration of Vectibix in combination with IFL should be avoided (see
section 4.5).
Vectibix in combination with bevacizumab and chemotherapy regimens
A randomized, open-label, multicentre study of 1,053 patients evaluated the efficacy of bevacizumab
and oxaliplatin- or irinotecan-containing chemotherapeutic regimens with and without Vectibix in the
first-line treatment of metastatic colorectal cancer. In an interim analysis based on 947 randomised
patients, shortened progression free survival time and increased deaths were observed in the patients
receiving Vectibix in combination with bevacizumab and chemotherapy. A greater frequency of
pulmonary embolism, infections (predominantly of dermatologic origin), diarrhoea, electrolyte
imbalances and dehydration was also observed in the treatment arms using Vectibix in combination
with bevacizumab and chemotherapy. An additional analysis of efficacy data by
KRAS
status did not
identify a subset of subjects who benefited from Vectibix in combination with oxaliplatin- or
irinotecan-based chemotherapy and bevacizumab. A trend towards worse survival was observed with
4
Vectibix in the wild-type
KRAS
subset of the oxaliplatin cohort, and a trend towards worse survival
was observed with Vectibix in the irinotecan cohort regardless of
KRAS
mutational status. Therefore,
Vectibix should not be administered in combination with bevacizumab containing chemotherapy (see
sections 4.5 and 5.1).
Vectibix in combination with oxaliplatin-based chemotherapy in metastatic colorectal cancer patients
Vectibix should not be administered in combination with oxaliplatin-containing chemotherapy to
mCRC patients with mutant
KRAS
tumours or for whom
KRAS
tumour status is unknown. In a phase 3
study (n = 1183, 656 subjects with wild-type
KRAS
and 440 subjects with mutant
KRAS
tumours)
evaluating panitumumab in combination with infusional 5-fluorouracil, leucovorin, and oxaliplatin
(FOLFOX) compared to FOLFOX alone as first-line therapy for mCRC, a shortened progression-free
survival and overall survival time were observed in patients with mutant
KRAS
tumours who received
panitumumab and FOLFOX (n = 221) vs. FOLFOX alone (n = 219).
Acute renal failure
Acute renal failure has been observed in patients who develop severe diarrhoea and dehydration.
No interaction studies have been performed.
Vectibix should not be administered in combination with IFL chemotherapy or with bevacizumab-
containing chemotherapy. A high incidence of severe diarrhoea was observed when panitumumab was
administered in combination with IFL (see section 4.4), and increased toxicity and deaths were seen
when panitumumab was combined with bevacizumab and chemotherapy (see sections 4.4 and 5.1).
Vectibix should not be administered to mCRC patients with mutant
KRAS
tumours or for whom
KRAS
status is unknown in combination with oxaliplatin-containing chemotherapy. A shortened progression-
free survival and overall survival time were observed in a clinical study in subjects with mutant
KRAS
tumours who received panitumumab and FOLFOX (see section 4.4).
Pregnancy
There are no adequate data from the use of Vectibix in pregnant women. Studies in animals have
shown reproductive toxicity (see section 5.3). The potential risk for humans is unknown. EGFR has
been implicated in the control of prenatal development and may be essential for normal organogenesis,
proliferation, and differentiation in the developing embryo. Therefore, Vectibix has the potential to
cause foetal harm when administered to pregnant women.
Human IgG is known to cross the placental barrier, and panitumumab may therefore be transmitted
from the mother to the developing foetus. In women of childbearing potential, appropriate
contraceptive measures must be used during treatment with Vectibix, and for 6 months following the
last dose. If Vectibix is used during pregnancy or if the patient becomes pregnant while receiving this
medicinal product, she should be advised of the potential risk for loss of the pregnancy or potential
hazard to the foetus.
Breast-feeding
It is unknown whether panitumumab is excreted in human breast milk. Because human IgG is secreted
into human milk, panitumumab might also be secreted. The potential for absorption and harm to the
infant after ingestion is unknown. It is recommended that women do not breast feed during treatment
with Vectibix and for 3 months after the last dose.
5
Fertility
Animal studies have shown reversible effects on the menstrual cycle and reduced female fertility in
monkeys (see section 5.3). Panitumumab may impact the ability of a woman to become pregnant.
No studies on the effects on the ability to drive and use machines have been performed.
If patients
experience treatment-related symptoms affecting their vision and/or ability to concentrate and react, it
is recommended that they do not drive or use machines until the effect subsides.
Based on an analysis of all clinical trial patients receiving Vectibix monotherapy (n = 1052), the most
commonly reported adverse reactions are skin reactions occurring in 93% of patients. These reactions
are related to the pharmacologic effects of Vectibix, and the majority are mild to moderate in nature
with 12% severe (grade 3 or higher, NCI-CTC). Commonly reported adverse reactions occurring in
≥ 20% of patients were gastrointestinal disorders [nausea (30%), diarrhoea (27%), and vomiting
(22%)]; general disorders [fatigue (35%)]; infections and infestations [paronychia (21%)]; and skin
and subcutaneous disorders [pruritus (53%), erythema (52%), dermatitis acneiform (51%), rash
(38%)].
Except where indicated, the data in the table below describe adverse reactions reported from clinical
studies in patients with metastatic colorectal carcinoma who received panitumumab as a single agent
(n = 1052). Within each frequency grouping, undesirable effects are presented in order of decreasing
seriousness.
Adverse reactions
MedDRA
system organ
class
Very common
(≥ 1/10)
Common
(≥ 1/100 to < 1/10)
Uncommon
(≥ 1/1000 to
< 1/100)
Rare
(≥1/10,000 to
<1/1000
Skin and
subcutaneous
tissue disorders
Dermatitis
acneiform
Rash
Exfoliative rash
Erythema
Skin exfoliation
Pruritus
Dry skin
Skin fissures
Acne
Palmar-plantar
erythrodysaesthesia
syndrome
Rash papular
Rash pruritic
Rash erythematous
Rash macular
Rash maculo-papular
Skin ulcer
Scab
Hypertrichosis
Alopecia
Onychoclasis
Nail disorder
(onycholysis)
Angioedema
1
Gastrointestinal
disorders
Diarrhoea
Nausea
Vomiting
Abdominal pain
Stomatitis
Constipation
Dry mouth
6











Adverse reactions
MedDRA
system organ
class
Very common
(≥ 1/10)
Common
(≥ 1/100 to < 1/10)
Uncommon
(≥ 1/1000 to
< 1/100)
Rare
(≥1/10,000 to
<1/1000
General
disorders and
administrative
site conditions
Fatigue
Pyrexia
Infusion-related
reaction
Mucosal inflammation
Chills
Chest discomfort
Infections and
infestations
Paronychia
Rash pustular
Eye infection
Eyelid infection
Cellulitis
Metabolism and
nutrition
disorders
Hypomagnesaemia
Hypocalcaemia
Hypokalaemia
Dehydration
Respiratory,
thoracic and
mediastinal
disorders
Dyspnoea
Cough
Pulmonary embolism
Epistaxis
Nasal dryness
Bronchospasm
Nervous system
disorders
Headache
Dizziness
Eye disorders
Conjunctivitis
Growth of eyelashes
Lacrimation increased
Ocular hyperaemia
Dry eye
Eye pruritus
Eyelid irritation
Eye irritation
Immune system
disorders
Hypersensitivity
Anaphylactic
reaction
Cardiac
disorders
Tachycardia
Cyanosis
Musculoskeletal
and connective
tissue disorders
Back pain
Vascular
disorders
Hypotension
Hypertension
Flushing
1
This adverse reaction was not reported in the monotherapy clinical studies (n=1052). Frequency was
derived from reports from all clinical studies performed with Vectibix (n = 4593)
The safety profile of panitumumab in patients whose tumour express
KRAS
wild-type (n = 394) was
generally consistent with the overall mCRC monotherapy set (n = 1052) described above. The only
differences were that nail disorder and hypomagnesaemia, were reported as very common (≥ 1/10) in
the
KRAS
wild-type arm whereas these adverse reactions were reported as common (≥ 1/100 to < 1/10)
in the overall mCRC monotherapy population and that stomatitis and acne were reported as common
in the
KRAS
wild-type versus very common in the overall mCRC monotherapy population. In
addition, bronchospasm, hypotension and hypertension were reported as uncommon (≥ 1/1000 to
< 1/100) in the overall mCRC monotherapy set and reported as common (≥ 1/100 to < 1/10) in the
KRAS
wild-type group.
7



















Gastrointestinal disorders
Diarrhoea when reported was mainly mild or moderate in severity. Two percent of patients with
KRAS
wild-type had diarrhoea reported as severe. There have been reports of acute renal failure in patients
who develop diarrhoea and dehydration (see section 4.4).
Infusion related reactions
In the setting of infusion-related reactions occurring within 24 hours of infusion, adverse reactions
including abdominal pain, anaphylactic reactions, angioedema, back pain, bronchospasm,
cardiorespiratory arrest, chest pain, chills, cyanosis, dyspnoea, flushing, hypertension, hypotension,
pyrexia, tachycardia and vomiting have been reported in clinical trials and in the post-marketing
setting. Across all clinical trials infusion-related reactions occurring within 24 hours of any infusion
were reported in 3% of Vectibix-treated patients, of which < 1% were severe (NCI-CTC grade 3 and
4). In the post-marketing setting, serious infusion reactions have been reported, including rare reports
with a fatal outcome.
A case of fatal angioedema occurred in a patient with recurrent and metastatic squamous cell
carcinoma of the head and neck treated with Vectibix in a clinical trial. The fatal event occurred after
re-exposure following a prior episode of angioedema; both episodes occurred greater than 24 hours
after administration (see sections 4.3 and 4.4). Hypersensitivity reactions occurring more than
24 hours after infusion have also been reported in the post-marketing setting.
For clinical management of infusion-related reactions, see section 4.4.
Skin and subcutaneous tissue disorders
Skin rash most commonly occurred on the face, upper chest, and back, but could extend to the
extremities. Subsequent to the development of severe skin and subcutaneous reactions, infectious
complications including sepsis, in rare cases leading to death, cellulitis and local abscesses requiring
incisions and drainage were reported. The median time to first symptom of dermatologic reaction was
10 days, and the median time to resolution after the last dose of Vectibix was 28 days.
Paronychial inflammation was associated with swelling of the lateral nail folds of the toes and fingers.
Dermatological reactions (including nail effects), observed in patients treated with Vectibix or other
EGFR inhibitors, are known to be associated with the pharmacologic effects of therapy. In the overall
monotherapy mCRC data set severe (grade 3 and grade 4) events included dermatitis acneiform (5%),
erythema (4%), rash (3%), pruritus (2%), exfoliative rash (1%), acne (1%), skin fissures (1%), skin
exfoliation (< 1%), dry skin (< 1%), skin ulcer (< 1%), scab (< 1%), rash erythematous (< 1%), rash
papular (< 1%), and rash maculo-papular < 1%). Paronychia was observed in 1% of patients with
Vectibix.
Vectibix in combination with other anti-cancer agents and/or monotherapy
Across all clinical trials, in combination with other anti-cancer agents and/or monotherapy, the most
serious adverse events associated with Vectibix treatment were pulmonary embolism, severe
dermatologic toxicity complicated by infectious sequelae and septic death, infusion-related reactions,
and hypomagnesaemia. Adverse reactions requiring discontinuation of Vectibix were infusion-related
reactions, severe skin toxicity and paronychia.
4.9
Doses up to 9 mg/kg have been tested in clinical trials. There have been reports of overdose at doses
up to approximately twice the recommended therapeutic dose. Adverse events observed included skin
toxicity, diarrhoea, dehydration and fatigue and were consistent with the safety profile at the
recommended dose.
8
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies, ATC code: L01XC08
Mechanism of action
Panitumumab is a recombinant, fully human IgG2 monoclonal antibody that binds with high affinity
and specificity to the human EGFR. EGFR is a transmembrane glycoprotein that is a member of a
subfamily of type I receptor tyrosine kinases including EGFR (HER1/c-ErbB-1), HER2, HER3, and
HER4. EGFR promotes cell growth in normal epithelial tissues, including the skin and hair follicle,
and is expressed on a variety of tumour cells.
Panitumumab binds to the ligand binding domain of EGFR and inhibits receptor autophosphorylation
induced by all known EGFR ligands. Binding of panitumumab to EGFR results in internalisation of
the receptor, inhibition of cell growth, induction of apoptosis, and decreased interleukin 8 and vascular
endothelial growth factor production.
The
KRAS
(Kirsten rat sarcoma 2 viral oncogene homologue) gene encodes a small, GTP-binding
protein involved in signal transduction. A variety of stimuli, including that from the EGFR activates
KRAS
which in turn stimulates other intracellular proteins to promote cell proliferation, cell survival
and angiogenesis.
Activating mutations in the
KRAS
gene occur frequently in a variety human tumours and have been
implicated in both oncogenesis and tumour progression.
Pharmacodynamic effects
In vitro
assays and
in vivo
animal studies have shown that panitumumab inhibits the growth and
survival of tumour cells expressing EGFR. No anti-tumour effects of panitumumab were observed in
human tumour xenografts lacking EGFR expression. The addition of panitumumab to radiation,
chemotherapy or other targeted therapeutic agents, in animal studies resulted in an increase in anti-
tumour effects compared to radiation, chemotherapy or targeted therapeutic agents alone.
Immunogenicity
Data on the development of anti-panitumumab antibodies has been evaluated using two different
immunoassays (an ELISA which detects high-affinity antibodies, and a Biosensor Immunoassay
which detects both high and low-affinity antibodies), results from these assays indicated that the
overall incidence of a post-dose anti-panitumumab antibody response was low. Pre-dose antibodies
were detected in 5 of 636 patients (< 1%) and 16/635 patients (2.5%) tested by the ELISA and
Biosensor Immunoassay respectively. Post-dose neutralising antibodies were detected in 1 of
447 patients (0.2%) and 7 of 447 patients (1.6%) tested by the ELISA and Biosensor Immunoassay
respectively. Compared with patients who did not develop antibodies, no relationship between the
presence of anti-panitumumab antibodies and pharmacokinetics, efficacy and safety has been
observed.
The detection of antibody formation is dependent on the sensitivity and specificity of the assay. The
observed incidence of antibody positivity in an assay may be influenced by several factors including
sample handling, concomitant medications and underlying disease, therefore, comparison of the
incidence of antibodies to other products may be misleading.
9
Clinical efficacy
The efficacy of Vectibix in patients with metastatic colorectal cancer (mCRC) who had disease
progression during or after prior chemotherapy was studied in a randomised controlled trial
(463 patients) and open-label, single-arm trials (384 patients). The safety of Vectibix in patients with
mCRC who received at least one dose of Vectibix was evaluated in 920 patients. Additional studies
were performed with Vectibix as a single agent in patients with other solid tumours and in
combination with chemotherapy with and without bevacizumab in patients with mCRC or in
combination with chemotherapy in patients with non-small cell lung cancer.
A multinational, randomised, controlled trial was conducted in 463 patients with EGFR-expressing
metastatic carcinoma of the colon or rectum after confirmed failure of oxaliplatin and irinotecan-
containing regimens. Patients were randomised 1:1 to receive Vectibix at a dose of 6 mg/kg given
once every two weeks plus best supportive care (not including chemotherapy) (BSC) or BSC alone.
Patients were treated until disease progression or unacceptable toxicity occurred. Upon disease
progression BSC alone patients were eligible to crossover to a companion study and receive Vectibix
at a dose of 6 mg/kg given once every two weeks.
Of 463 patients, 63% were male. The median age was 62 years (range 27 to 83), and 99% were
Caucasian. Three hundred and ninety-six (86%) patients had a baseline ECOG Performance Status of
0 or 1. Sixty-seven percent of patients had colon cancer and 33% had rectal cancer.
The primary endpoint was progression-free survival (PFS). In an analysis adjusting for potential bias
from unscheduled assessments, the rate of disease progression or death in patients who received
Vectibix was reduced by 40% relative to patients that received BSC [Hazard Ratio = 0.60, (95% CI
0.49, 0.74), stratified log-rank p < 0.0001]. There was no difference seen in median PFS times as more
than 50% of patients progressed in both treatment groups before the first scheduled visit. The
progression-free survival rates at the first scheduled visit (week 8) were 45.5% on Vectibix plus BSC
and 24.6% on BSC alone, a difference of 20.9% [95% CI: 12.4, 29.4]. No difference was seen in
overall survival. This may be due to patients receiving panitumumab after progression among those
randomized to BSC. Tumour response according to modified-RECIST criteria was determined by
central review. Overall, 9.5% [95% CI: 6.1, 14.1] Vectibix plus BSC patients, and 0%
[95% CI: 0.0, 1.6] BSC alone patients had a confirmed objective response (partial response), with
stable disease in 26% and 10% patients, respectively. Among the 176 patients who received Vectibix
after progression on BSC alone, the response rate (investigator assessment) was 11.4% (95%
CI: 7.1, 17.0).
10
PFS – All Patients
100%
90%
Median
i
n Weeks
Treatment Group
E
vents / N (%)
80%
Vectibix+BSC 193 / 231 ( 84 )
8.0
70%
BSC Alone
208 / 232 ( 90 )
8.0
60%
50%
Hazard ratio = 0.60
(95% CI: 0.49, 0.74)
Stratified log-rank test p<0.0001
40%
30%
20%
10%
0%
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 52
Subjects at risk:
Vectibix+BSC
BSC Alone
Weeks
231 228 221 216 212 85 84 65 64 41 40 40 40 22 19 19 19 8 8 8 7 2 2 1 1 0
232 230 225 220 216 39 39 20 20 11 10 7 7 4 4 4 4 3 2 2 2 1 1 1 1 0
Unscheduled tumour assessments were moved to the nearest scheduled timepoint
The relationship between
KRAS
mutation status determined in archived paraffin embedded tumour
tissue and clinical outcome was evaluated in a retrospective analysis.
Tumour samples obtained from the primary resection of colorectal cancer were analysed for the
presence of the seven most common activating mutations in the codon 12 and 13 (Gly12Asp,
Gly12Ala, Gly12Val, Gly12Ser, Gly12Arg, Gly12Cys, and Gly13Asp) of the
KRAS
gene by using an
allele-specific polymerase chain reaction. 427 (92%) patients were evaluable for
KRAS
status of which
184 had mutations. In an analysis adjusting for potential bias from unscheduled assessments the
hazard ratio for PFS was 0.49 (95% CI: 0.37-0.65) in favour of panitumumab in the
KRAS
wild-type
group and 1.07 (95% CI: 0.77-1.48) in the
KRAS
mutant group. The difference in median PFS in the
KRAS
wild-type group was 8 weeks. The progression-free survival rates at the first scheduled visit
(week 8) in the
KRAS
wild-type group were 59.7% on Vectibix plus BSC and 21.0% on BSC alone, a
difference of 38.7% [95% CI: 27.4, 50.0]. The difference in median PFS in the
KRAS
mutant group
was 0 weeks. The progression-free survival rates at the first scheduled visit (week 8) in the
KRAS
mutant group were 21.4% on Vectibix plus BSC and 28.0% on BSC alone, a difference of -6.6%
[95% CI: -19.0, 5.9]. There were no differences in overall survival seen in either group. In the
KRAS
wild-type group the response rate was 17% for panitumumab and 0% for BSC. In the
KRAS
mutant
group there were no responses in either treatment arm. Stable disease rates in the
KRAS
wild-type
group were 34% for panitumumab and 12% for BSC. The stable disease rates in the
KRAS
mutant
group were 12% for panitumumab and 8% for BSC. Response rate (investigator assessment) in
patients that crossed over to panitumumab after progression on BSC alone was 22%
(95% CI: 14.0, 31.9) for those with
KRAS
wild-type tumours and 0% (95% CI: 0.0, 4.3) for those with
mutant
KRAS
tumours.
11



















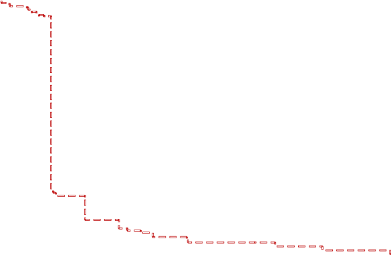







































































































































































PFS – Patients with mutant and wild type KRAS
Wild Type
KRAS
100%
Median
in
Weeks
Vectibix+BSC 115 / 124 ( 93 ) 16.0
BSC Alone
E
vents / N (%)
90%
114 / 119 ( 96 )
8.0
80%
70%
Hazard ratio = 0.49
(95% CI: 0.37, 0.65)
Stratified log-rank test p<0.0001
60%
50%
40%
30%
20%
10%
0%
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 52
Subjects at risk:
Vectibix+BSC
BSC Alone
Weeks
124 122 116 114 114 69 69 58 58 45 44 44 44 24 20 20 20 13 13 13 12 7 7 6 6 4
119 118 116 116 114 19 19 15 15 11 11 9 9 6 6 6 6 5 4 3 3 2 2 2 2 1
Unscheduled tumour assessments were moved to the nearest scheduled timepoint
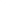
Mutant
KRAS
Median
in
Weeks
100%
Treatment Group
E
vents / N (%)
Vectibix+BSC
76 / 84 ( 90 )
8.0
90%
BSC Alone
95 / 100 ( 95 )
8.0
80%
70%
60%
Hazard ratio = 1.07
(95% CI: 0.77, 1.48)
50%
40%
30%
20%
10%
0%
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 52
Subjects at risk:
Vectibix+BSC
BSC Alone
Weeks
84 84 82 81 77 10 9 6 6 5 5 5 5 4 4 4 4 2 2 2 2 2 2 1 1 1
100 99 97 91 90 22 22 10 10 8 7 5 5 4 4 4 4 4 4 4 4 3 2 2 2 2
Unscheduled tumour assessments were moved to the nearest scheduled timepoint
The PACCE study: In this randomised, open label, controlled clinical trial, chemotherapy (oxaliplatin
or irinotecan) and bevacizumab were given with and without panitumumab in the first line treatment
of patients with metastatic colorectal cancer (n = 1053 [n = 823 oxaliplatin cohort, n = 230 irinotecan
cohort]). Panitumumab treatment was discontinued due to a statistically significant reduction in PFS in
patients receiving panitumumab observed in an interim analysis.
The major study objective was comparison of PFS in the oxaliplatin cohort. In the final analysis, the
hazard ratio for PFS was 1.27 (95% CI: 1.06, 1.52). Median PFS was 10.0 (95% CI: 8.9, 11.0) and
11.4 (95% CI: 10.5, 11.9) months in the panitumumab and the non-panitumumab arm, respectively.
12
Treatment Group































































































































































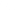








































































































































































































There was an increase in mortality in the panitumumab arm. The hazard ratio for overall survival was
1.43 (95% CI: 1.11, 1.83). Median overall survival was 19.4 (95% CI: 18.4, 20.8) and 24.5 (95% CI:
20.4, 24.5) in the panitumumab arm and the non-panitumumab arm.
An additional analysis of efficacy data by
KRAS
status did not identify a subset of subjects who
benefited from panitumumab in combination with oxaliplatin- or irinotecan based chemotherapy and
bevacizumab. For the wild-type
KRAS
subset of the oxaliplatin cohort, the hazard ratio for PFS was
1.36 with 95% CI: 1.04-1.77. For the mutant
KRAS
subset, the hazard ratio for PFS was 1.25 with 95%
CI: 0.91-1.71. A trend for OS favouring the control arm was observed in the wild-type
KRAS
subset of
the oxaliplatin cohort (hazard ratio = 1.89; 95% CI: 1.30, 2.75). A trend towards worse survival was
also observed with panitumumab in the irinotecan cohort regardless of
KRAS
mutational status.
Overall, panitumumab treatment combined with chemotherapy and bevacizumab is associated with an
unfavourable benefit-to-risk profile irrespective of tumour
KRAS
mutational status.
This medicinal product has been authorised under a “conditional approval” scheme. This means that
further evidence on this medicinal product is awaited, in particular data are required to confirm the
effect in patients with wild-type
KRAS
tumours which is currently supported by a retrospective
analysis. Further evidence is also awaited regarding the effect of panitumumab in combination with
chemotherapy on PFS in patients with wild-type
KRAS
tumours. Studies investigating this effect are
currently ongoing. The European Medicines Agency (EMEA) will review new information on the
product every year and this SPC will be updated as necessary.
5.2 Pharmacokinetic properties
Vectibix administered as a single agent or in combination with chemotherapy exhibits nonlinear
pharmacokinetics.
Following a single-dose administration of panitumumab as a 1-hour infusion, the area under the
concentration-time curve (AUC) increased in a greater than dose-proportional manner and clearance
(CL) of panitumumab decreased from 30.6 to 4.6 ml/day/kg as the dose increased from 0.75 to
9 mg/kg. However, at doses above 2 mg/kg, the AUC of panitumumab increases in an approximately
dose-proportional manner.
Following the recommended dose regimen (6 mg/kg given once every 2 weeks as a 1-hour infusion),
panitumumab concentrations reached steady-state levels by the third infusion with mean (± SD) peak
and trough concentrations of 213 ± 59 and 39 ± 14 mcg/ml, respectively. The mean (± SD) AUC0-tau
and CL were 1306 ± 374 mcg•day/ml and 4.9 ± 1.4 ml/kg/day, respectively. The elimination half-life
was approximately 7.5 days (range: 3.6 to 10.9 days).
A population pharmacokinetic analysis was performed to explore the potential effects of selected
covariates on panitumumab pharmacokinetics. Results suggest that age (21-88), gender, race, hepatic
function, renal function, chemotherapeutic agents, and EGFR membrane staining intensity (1+, 2+,
3+) in tumour cells had no apparent impact on the pharmacokinetics of panitumumab.
No clinical studies have been conducted to examine the pharmacokinetics of panitumumab in patients
with renal or hepatic impairment.
5.3 Preclinical safety data
Adverse reactions seen in animals at exposure levels similar to clinical exposure levels and with
possible relevance to clinical use were as follows:
Skin rash and diarrhoea were the major findings observed in repeat-dose toxicity studies of up to
26 weeks duration in cynomolgus monkeys. These findings were observed at doses approximately
equivalent to the recommended human dose and were reversible upon termination of administration of
panitumumab. The skin rash and diarrhoea observed in monkeys are considered related to the
13
pharmacological action of panitumumab and are consistent with the toxicities observed with other
anti-EGFR inhibitors.
Studies to evaluate the mutagenic and carcinogenic potential of panitumumab have not been
performed.
Animal studies are insufficient with respect to embryo-foetal development since foetal panitumumab
exposure levels were not examined. Panitumumab has been shown to cause foetal abortions and/or
foetal deaths in cynomolgus monkeys when administered during the period of organogenesis at doses
approximately equivalent to the recommended human dose.
Formal male fertility studies have not been conducted; however, microscopic evaluation of male
reproductive organs from repeat-dose toxicity studies in cynomolgus monkeys at doses up to
approximately 5-fold the human dose on a mg/kg basis, revealed no differences compared to control
male monkeys. Fertility studies conducted in female cynomolgus monkeys showed that panitumumab
may produce prolonged menstrual cycle and/or amenorrhea and reduced pregnancy rate which
occurred at all doses evaluated.
No pre- and post-natal development animal studies have been conducted with panitumumab. All
patients should be advised regarding the potential risk of panitumumab on pre- and post-natal
development prior to initiation of Vectibix therapy.
6.
6.1
List of excipients
Sodium chloride
Sodium acetate trihydrate
Acetic acid, glacial (for pH-adjustment)
Water for injections.
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in
section 6.6.
6.3 Shelf life
3 years.
Vectibix does not contain any antimicrobial preservative or bacteriostatic agent. The product should be
used immediately after dilution. If not used immediately, in-use storage times and conditions prior to
use are the responsibility of the user and should be no longer than 24 hours at 2°C to 8°C. Do not
freeze diluted solution.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C).
Do not freeze.
Store in the original carton in order to protect from light.
For storage conditions of the diluted medicinal product, see section 6.3.
6.5
Nature and contents of container
Single-use vial (type I glass) with an elastomeric stopper, aluminium seal and flip-off plastic cap.
14
One vial contains: 100 mg of panitumumab in 5 ml, 200 mg panitumumab in 10 ml, or 400 mg
panitumumab in 20 ml of concentrate for solution for infusion.
Pack of 1 vial containing 5 ml.
Pack of 1 vial containing 10 ml.
Pack of 1 vial containing 20 ml.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
. Do not administer Vectibix if discolouration is
observed. Withdraw the necessary amount of Vectibix for a dose of 6 mg/kg. Dilute in a total volume
of 100 ml. The final concentration should not exceed 10 mg/ml. Doses higher than 1000 mg should be
diluted in 150 ml 0.9% sodium chloride injection (see section 4.2). The diluted solution should be
mixed by gentle inversion, do not shake.
No incompatibilities have been observed between Vectibix and 0.9% sodium chloride injection in
polyvinyl chloride bags or polyolefin bags.
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Amgen Europe B.V.
Minervum 7061
NL-4817 ZK Breda
The Netherlands
8.
EU/1/07/423/001
EU/1/07/423/002
EU/1/07/423/003
9.
Date of first authorisation: 3 December 2007
Date of last renewal: 17 March 2011
Detailed information on this medicine is available on the website of the European Medicines Agency
15
Vectibix should be diluted in 0.9% sodium chloride injection by healthcare professional using aseptic
technique.
Do not shake or vigorously agitate the vial
ANNEX II
A.
MANUFACTURER OF THE BIOLOGICAL ACTIVE
SUBSTANCE AND MANUFACTURING AUTHORISATION
HOLDER RESPONSIBLE FOR BATCH RELEASE
B.
CONDITIONS OF THE MARKETING AUTHORISATION
C.
16
A. MANUFACTURER OF THE BIOLOGICAL ACTIVE SUBSTANCE AND
MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturer of the biological active substance
Amgen Inc.
6701 Kaiser Drive
Fremont, CA 94555
USA
Name and address of the manufacturer responsible for batch release
Amgen Europe B.V.
Minervum 7061
NL-4817 ZK Breda
The Netherlands
B. CONDITIONS OF THE MARKETING AUTHORISATION
•
Medicinal p roduct su bject t o r estricted medical p rescription ( See An nex I : S ummary o f P roduct
Characteristics, section 4.2).
•
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND
EFFECTIVE USE OF THE MEDICINAL PRODUCT
Not applicable
•
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, as described in version 5 presented in
Module 1.8.1. of the Marketing Authorisation Application, is in place and functioning before and
whilst the product is on the market.
Risk Management Plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in
the Pharmacovigilance Plan, as agreed in version 5 of the Risk Management Plan (RMP) presented in
Module 1.8.2. of the Marketing Authorisation Application and any subsequent updates of the RMP
agreed by the CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, the
updated R MP s hould be s ubmitted a t t he sa me t ime a s t he n ext P eriodic S afety Up date R eport
(PSUR).
In addition, an updated RMP should be submitted:
•
When n ew i nformation i s r eceived t hat may i mpact o n t he cu rrent S afety S pecification,
Pharmacovigilance Plan or risk minimisation activities
•
Within 6 0 d ays of a n i mportant ( pharmacovigilance or r isk minimisation) m ilestone be ing
reached
•
At the request of the European Medicines Agency
17
C. SPECIFIC OBLIGATIONS TO BE FULFILLED BY THE MARKETING
AUTHORISATION HOLDER
The Marketing Authorisation Holder shall complete the following programme of studies within the
specified t ime f rame, t he results o f which sh all f orm t he b asis of t he an nual r eassessment o f t he
benefit/risk profile:
•
To submit the clinical study summary report of the SPIRITT study including the safety-efficacy
analysis in relation with
KRAS
by Q1-2011
•
To complete a confirmatory trial examining panitumumab monotherapy in licensed indication.
In particular to:
- provide the clinical study report of the primary data analysis from the 20080763 study by
Q4-2012
•
To resolve the uncertainties about
KRAS
testing by end May 2012 by:
-
collecting information about the range of diagnostic tests conducted in clinical practice
and their performance
-
collecting data on and evaluating the compliance of physicians with the recommend use
of Vectibix in confirmed cases of wild-type tumours
18
ANNEX III
LABELLING AND PACKAGE LEAFLET
19
A. LABELLING
20
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
CARTON
1.
Vectibix 20 mg/ml concentrate for solution for infusion
Panitumumab
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each vial contains 100 mg of panitumumab.
Each vial contains 200 mg of panitumumab.
Each vial contains 400 mg of panitumumab.
3.
LIST OF EXCIPIENTS
Sodium chloride, sodium acetate trihydrate, acetic acid (glacial), water for injections.
4.
5 ml of concentrate for solution for infusion.
10 ml of concentrate for solution for infusion.
20 ml of concentrate for solution for infusion.
x1
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
For intravenous use.
Read the package leaflet before use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
Do not shake.
8.
EXPIRY DATE
EXP
21


























9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator.
Do not freeze.
Store in the original carton in order to protect from light.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
11.
Amgen Europe B.V.
Minervum 7061
NL-4817 ZK Breda
The Netherlands
12.
EU/1/07/423/001
EU/1/07/423/002
EU/1/07/423/003
13.
BATCH NUMBER
Lot
14.
GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15.
INSTRUCTIONS ON USE
16.
INFORMATION IN BRAILLE
Justification for not including Braille accepted.
22























MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
VIAL LABEL
1.
Vectibix 20 mg/ml sterile concentrate
Panitumumab
IV
2.
METHOD OF ADMINISTRATION
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
100 mg/5 ml
200 mg/10 ml
400 mg/20 ml
6.
OTHER
Amgen Europe B.V.
23




















B. PACKAGE LEAFLET
24
PACKAGE LEAFLET: INFORMATION FOR THE USER
Vectibix 20 mg/ml concentrate for solution for infusion
panitumumab
Read all of this leaflet carefully before you start using this medicine.
•
Keep this leaflet. You may need to read it again.
•
If you have any further questions, ask your doctor.
•
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
•
If any of the side effects get serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor.
In this leaflet:
1. What Vectibix is and what it is used for
2. Before you use Vectibix
3. How to use Vectibix
4. Possible side effects
5.
How to store Vectibix
6. Further information
1.
WHAT VECTIBIX IS AND WHAT IT IS USED FOR
Vectibix is used in the treatment of metastatic colorectal carcinoma (cancer of the bowel) after failure
of chemotherapy (medicines used to treat cancer) treatment.
Vectibix is for use in adults 18 years and over.
Vectibix contains the active substance panitumumab, which belongs to a group of medicines called
monoclonal antibodies. Monoclonal antibodies are proteins, which specifically recognise and attach
(bind) to other unique proteins in the body.
Panitumumab recognises and binds specifically to a protein known as epidermal growth factor
receptor (EGFR), which is found on the surface of some cancer cells. When growth factors (other
body proteins) attach to the EGFR, the cancer cell is stimulated to grow and divide. Panitumumab
binds onto the EGFR and prevents the cancer cell from receiving the messages it needs for growth and
division.
2.
BEFORE YOU USE
VECTIBIX
Do not use Vectibix
•
if you have ever had a severe or life-threatening allergic (hypersensitivity) reaction to
panitumumab or any of the other ingredients of Vectibix.
•
if you have previously had or have evidence of interstitial pneumonitis (swelling of the lungs
causing coughing and difficulty breathing) or pulmonary fibrosis (scarring and thickening in the
lungs with shortness of breath).
25
Take special care with Vectibix
Your doctor will check your blood levels of several substances such as magnesium, and other
electrolyte levels such as calcium and potassium in your blood before you start Vectibix treatment. If
these levels are too low, your doctor may prescribe you appropriate supplements.
During treatment with Vectibix
You may experience dermatologic toxicities (skin reactions), if these worsen or become intolerable
please tell your doctor or nurse immediately.
It is recommended that you limit sun exposure whilst receiving Vectibix and if you are experiencing
skin reactions as sunlight can worsen these. Wear sunscreen and a hat if you are going to be exposed
to sunlight.
Your doctor will ask you to come in for tests to monitor hypomagnesaemia (low magnesium levels in
the blood) and hypocalcaemia (low calcium levels in the blood) periodically during your treatment,
and for up to 8 weeks after you have finished your treatment.
Using other medicines
Please tell your doctor or pharmacist if you are taking or have recently taken any other medicines,
including medicines obtained without a prescription.
Pregnancy and breast-feeding
Vectibix has not been tested in pregnant women. It is important to tell your doctor if you are pregnant;
think you may be pregnant; or plan to get pregnant. Vectibix could affect your ability to stay pregnant.
If you are a woman of child bearing potential, you should use suitable methods of contraception
during treatment with Vectibix and for 6 months after the last dose.
Do not breast-feed your baby during treatment with Vectibix and for 3 months after the last dose.
Ask your doctor or pharmacist for advice before taking any medicine.
Driving and using machines
No studies on the effects on the ability to drive and use machines have been performed. You should
speak with your doctor before driving or using machines, as some side effects may impair your ability
to do so safely.
3.
HOW TO USE VECTIBIX
Vectibix will be administered in a healthcare facility under the supervision of a doctor experienced in
the use of anti-cancer medicines.
Vectibix is administered intravenously (into a vein) with an infusion pump (a device that gives a slow
injection).
The recommended dose of Vectibix is 6 mg/kg (milligrams per kilogram of body weight) given once
every two weeks. The treatment will usually be given over a period of approximately 60 minutes.
26
4.
POSSIBLE SIDE EFFECTS
Like all medicines, Vectibix can cause side effects, although not everybody gets them.
Very common side effects
(seen in more than 1 in 10 people who take Vectibix) were:
•
acne-like rash; acne; pruritus (itching); erythema (redness of skin); rash; skin exfoliation
(flaking skin); dry skin; skin fissures (cracks in the skin); exfoliating rash (flaking rash);
•
diarrhoea; nausea; vomiting; abdominal pain; constipation;
•
stomatitis (chapped lips, mouth ulcers and cold sores);
•
fatigue (extreme tiredness);
•
pyrexia (fever or high temperature);
•
paronychia (nail infection);
•
cough; dyspnoea (breathing difficulties).
Common side effects
(seen in more than 1, but less than 10 in 100 people taking Vectibix) were:
•
infusion type reactions which may include signs and symptoms such as abdominal pain, back
pain, breathing difficulties, chest pain, flushing, rapid heart rate,; hypotension (low blood
pressure); hypertension (high blood pressure);, vomiting; chills; new onset of facial swelling
and/or swelling of the mouth; and/or pyrexia (fever or high temperature);
•
hand-foot syndrome (redness and swelling of palms of hands or soles of feet);
•
onycholysis (loosening of the nails); nail disorder;
•
rash pustular (skin rash with pus-filled blisters);
•
eye infection; eyelid infection;
•
cellulitis (spreading infection below the skin);
•
hypomagnesaemia (low magnesium levels in the blood);
•
hypocalcaemia (low calcium levels in the blood);
•
hypokalaemia (low potassium levels in the blood);
•
dehydration;
•
nasal dryness; epistaxis (nose bleed);
•
headache; dizziness;
•
rash papular (bumpy rash); rash pruritic (itchy rash); rash erythematous (red skin rash); rash
macular (spotty rash); rash maculo-papular (rash with bumps and spots); skin ulcer; scab;
•
conjunctivitis (eye inflammation); growth of eyelashes and lacrimation increased (flow of
tears); ocular hyperaemia (redness of the eye); dry eye; eye pruritus (itchy eyes); eyelid
irritation; eye irritation;
•
pulmonary embolism (blood clot in the lung);
•
mucosal inflammation (inflammation of the mouth); dry mouth;
•
onychoclasis (breaking of the nails);
•
hypertrichosis (excess hair growth); alopecia (hair loss).
Uncommon side effects
(seen in less than 1 in 100, but more than 1 in 1000 people taking Vectibix)
were:
•
bronchospasm (constriction of the airways);
•
anaphylactic reactions (severe allergic reaction);
•
flushing; hypotension (low blood pressure); hypertension (high blood pressure);
•
cyanosis (blue coloration of the skin and mucous membranes).
Rare side effects
(seen in less than 1 in 1000, but more than 1 in 10,000 people taking Vectibix) were:
•
angioedema (swelling of the mouth, face and throat causing difficulty in breathing).
Infusion-type reactions, which may include signs and symptoms such as chills, new onset of facial
swelling, breathing difficulties, vomiting and/or fever or pyrexia (high temperature) may appear
several hours or days after an infusion. If any of these side effects gets serious
,
please tell your doctor.
27
If any of the side effects get serious
, or if you notice any side effects not listed in this leaflet, please
tell your doctor.
Important information about some of the ingredients of Vectibix
This medicinal product contains 0.150 mmol sodium (which is 3.45 mg sodium) per ml of concentrate.
To be taken into consideration by patients on a controlled sodium diet.
5.
HOW TO STORE VECTIBIX
Vectibix will be stored in the healthcare facility where it is used.
Keep out of the reach and sight of children.
Store in a refrigerator (2°C – 8°C).
Do not freeze.
Store in the original carton in order to protect from light.
Do not use Vectibix after the expiry date which is stated on the label and carton after EXP. The expiry
date refers to the last day of that month.
Medicines should not be disposed of via wastewater or household waste. Ask your pharmacist how to
dispose of medicines no longer required. These measures will help to protect the environment.
6.
FURTHER INFORMATION
What Vectibix contains
The active substance is panitumumab 20 mg/ml.
The other ingredients of Vectibix are sodium chloride, sodium acetate trihydrate, acetic acid (glacial)
and water for injections.
What Vectibix looks like and contents of the pack
Vectibix is a colourless liquid that may contain visible particles and is supplied in a vial. Each pack
contains one vial of either 5 ml, 10 ml or 20 ml of concentrate.
Not all pack sizes may be marketed.
Marketing Authorisation Holder and Manufacturer
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
The Netherlands
For any information about this medicinal product, please contact the local representative of the
Marketing Authorisation Holder.
België/Belgique/Belgien
s.a. Amgen n.v.
Tel/Tél: +32 (0)2 7752711
Luxembourg/Luxemburg
s.a. Amgen
Belgique/Belgien
Tel/Tél: +32 (0)2 7752711
28
България
Амджен България ЕООД
Тел.: +359 (0)2 805 7020
Magyarország
Amgen Kft.
Tel.: +36 1 35 44 700
Česká republika
Amgen s.r.o
Tel: +420 2 21 773 500
Malta
Amgen B.V.
The Netherlands
Tel: +31 (0)76 5732500
Danmark
Amgen filial af Amgen AB, Sverige
Tlf: +45 39617500
Nederland
Amgen B.V.
Tel: +31 (0)76 5732500
Deutschland
AMGEN GmbH
Tel: +49 (0)89 1490960
Norge
Amgen AB
Tlf: +47 23308000
Eesti
Amgen Switzerland AG Eesti filiaal
Tel: +372 5125 501
Österreich
Amgen GmbH
Tel: +43 (0)1 50 217
Ελλάδα
Amgen Ελλάς Φαρμακευτικά ΕΠΕ.
Τηλ.: +30 210 3447000
Polska
Amgen Sp. z o.o.
Tel.: +48 22 581 3000
España
Amgen S.A.
Tel: +34 93 600 19 00
Portugal
AMGEN Biofarmacêutica, Lda.
Tel: +351 21 4220550
France
Amgen S.A.S
Tél: +33 (0)1 40 88 27 00
România
Amgen România SRL
Tel: +4021 527 3000
Ireland
Amgen Limited
United Kingdom
Tel:
+44 (0)1223 420305
Slovenská republika
Amgen Switzerland AG Slovakia
Tel: +421 33 321 13 22
Ísland
Vistor hf.
Sími: +354 535 7000
Slovenija
AMGEN zdravila d.o.o.
Tel: +386 1 585 1767
Italia
Amgen Dompé S.p.A.
Tel: +39 02 6241121
Suomi/Finland
Amgen AB, sivuliike Suomessa/Amgen AB, filial
i Finland
Puh/Tel: +358 (0)9 54900500
Kύπρος
Papaellinas & Co Ltd
Τηλ: +357 22741 741
Sverige
Amgen AB
Tel: +46 (0)8 6951100
Latvija
Amgen Switzerland AG Rīgas filiāle
Tel: +371 29284 807
United Kingdom
Amgen Limited
Tel: +44 (0)1223 420305
29
Lietuva
Amgen Switzerland AG Vilniaus filialas
Tel: +370 6983 6600
This leaflet was last approved in
Detailed information on this medicine is available on the European Medicines Agency web site:
This medicine has been given “conditional approval”.
This means that there is more evidence to come about this medicine.
The European Medicines Agency (EMA) will review new information on the medicine every year and
this leaflet will be updated as necessary.
---------------------------------------------------------------------------------------------------------------------------
The following information is intended for medical or healthcare professionals only:
. Do not administer Vectibix if discolouration is
observed. Withdraw the necessary amount of Vectibix for a dose of 6 mg/kg. Dilute in a total volume
of 100 ml. Doses higher than 1000 mg should be diluted in 150 ml 0.9% sodium chloride injection.
The final concentration should not exceed 10 mg/ml. The diluted solution should be mixed by gentle
inversion, do not shake.
The infusion line should be flushed with sodium chloride solution before and after Vectibix
administration to avoid mixing with other medicinal products or IV solutions.
Vectibix must be administered as an intravenous infusion via an infusion pump, using a low protein
binding 0.2 or 0.22 micrometer in-line filter, through a peripheral line or indwelling catheter. The
recommended infusion time is approximately 60 minutes. Doses higher than 1000 mg should be
infused over approximately 90 minutes.
No incompatibilities have been observed between Vectibix and 0.9% sodium chloride injection in
polyvinyl chloride bags or polyolefin bags.
30
Vectibix should be diluted in 0.9% sodium chloride injection by healthcare professional using aseptic
technique.
Do not shake or vigorously agitate the vial
Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/vectibix.html
Copyright © 1995-2021 ITA all rights reserved.