










































ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
1.
Zarzio 30 MU/0.5 ml solution for injection or infusion in pre-filled syringe
2.
Each ml of solution contains 60 million units (MU) [equivalent to 600 micrograms (μg)] filgrastim*.
Each pre-filled syringe contains 30 MU (equivalent to 300 μg) filgrastim in 0.5 ml.
* recombinant methionylated human granulocyte-colony stimulating factor (G-CSF) produced in
E. coli
by recombinant DNA technology.
Excipient: Each ml of solution contains 50 mg sorbitol (E420).
For a full list of excipients, see section 6.1.
3.
Solution for injection or infusion in pre-filled syringe
Clear, colourless to slightly yellowish solution.
4.
-
Reduction in the duration of neutropenia and the incidence of febrile neutropenia in patients
treated with established cytotoxic chemotherapy for malignancy (with the exception of chronic
myeloid leukaemia and myelodysplastic syndromes) and reduction in the duration of
neutropenia in patients undergoing myeloablative therapy followed by bone marrow
transplantation considered to be at increased risk of prolonged severe neutropenia.
The safety and efficacy of filgrastim are similar in adults and children receiving cytotoxic
chemotherapy.
-
Mobilisation of peripheral blood progenitor cells (PBPC).
-
In children and adults with severe congenital, cyclic, or idiopathic neutropenia with an absolute
neutrophil count (ANC) of ≤ 0.5 x 10
9
/l, and a history of severe or recurrent infections, long
term administration of filgrastim is indicated to increase neutrophil counts and to reduce the
incidence and duration of infection-related events.
-
Treatment of persistent neutropenia (ANC ≤ 1.0 x 10
9
/l) in patients with advanced HIV
infection, in order to reduce the risk of bacterial infections when other therapeutic options are
inappropriate.
Filgrastim therapy should only be given in collaboration with an oncology centre which has
experience in granulocyte-colony stimulating factor (G-CSF) treatment and haematology and has the
necessary diagnostic facilities.
2
The mobilisation and apheresis procedures should be performed in collaboration with an
oncology-haematology centre with acceptable experience in this field and where the monitoring of
haematopoietic progenitor cells can be correctly performed.
Zarzio is available in strengths of 30 MU/0.5 ml and 48 MU/0.5 ml.
Established cytotoxic chemotherapy
The recommended dose of filgrastim is 0.5 MU/kg/day (5 μg/kg/day). The first dose of filgrastim
should not be administered less than 24 hours following cytotoxic chemotherapy.
Daily dosing with filgrastim should continue until the expected neutrophil nadir is passed and the
neutrophil count has recovered to the normal range. Following established chemotherapy for solid
tumours, lymphomas, and lymphoid leukaemias, it is expected that the duration of treatment required
to fulfil these criteria will be up to 14 days. Following induction and consolidation treatment for acute
myeloid leukaemia the duration of treatment may be substantially longer (up to 38 days) depending on
the type, dose and schedule of cytotoxic chemotherapy used.
In patients receiving cytotoxic chemotherapy, a transient increase in neutrophil counts is typically seen
1 - 2 days after initiation of filgrastim therapy. However, for a sustained therapeutic response,
filgrastim therapy should not be discontinued before the expected nadir has passed and the neutrophil
count has recovered to the normal range. Premature discontinuation of filgrastim therapy, prior to the
time of the expected neutrophil nadir, is not recommended.
Patients treated with myeloablative therapy followed by bone marrow transplantation
The recommended starting dose of filgrastim is 1.0 MU/kg/day (10 μg/kg/day). The first dose of
filgrastim should not be administered less than 24 hours following cytotoxic chemotherapy and within
24 hours of bone marrow infusion.
Dose adjustments:
Once the neutrophil nadir has been passed, the daily dose of filgrastim should be
titrated against the neutrophil response as follows:
Absolute neutrophil count
Filgrastim dose adjustment
ANC > 1.0 x 10
9
/l for 3 consecutive days
Reduce to 0.5 MU/kg/day (5 μg/kg/day)
Then, if ANC remains > 1.0 x 10
9
/l for
3 more consecutive days
Discontinue filgrastim
If the ANC decreases to < 1.0 x 10
9
/l during the treatment period, the dose of filgrastim
should be re-escalated according to the above steps
Mobilisation of PBPC
Patients undergoing myelosuppressive or myeloablative therapy followed by autologous PBPC
transplantation
The recommended dose of filgrastim for PBPC mobilisation when used alone is
1.0 MU/kg/day (10 μg/kg/day) for 5 - 7 consecutive days. Timing of leukapheresis:
1 or 2 leukaphereses on days 5 and 6 are often sufficient. In other circumstances, additional
leukaphereses may be necessary. Filgrastim dosing should be maintained until the last leukapheresis.
The recommended dose of filgrastim for PBPC mobilisation after myelosuppressive chemotherapy is
0.5 MU/kg/day (5 μg/kg/day) given daily from the first day after completion of chemotherapy until the
expected neutrophil nadir is passed and the neutrophil count has recovered to the normal range.
Leukapheresis should be performed during the period when the ANC rises from < 0.5 x 10
9
/l to
> 5.0 x 10
9
/l. For patients who have not had extensive chemotherapy, one leukapheresis is often
sufficient. In other circumstances, additional leukaphereses are recommended.
There are no prospectively randomised comparisons of the two recommended mobilisation methods
(filgrastim alone or in combination with myelosuppressive chemotherapy) within the same patient
population. The degree of variation between individual patients and between laboratory assays of
3







CD34
+
cells means that direct comparison between different studies is difficult. It is therefore difficult
to recommend an optimum method. The choice of mobilisation method should be considered in
relation to the overall objectives of treatment for an individual patient.
Normal donors prior to allogeneic PBPC transplantation
For PBPC mobilisation in normal donors prior to allogeneic PBPC transplantation, filgrastim should
be administered at 1.0 MU/kg/day (10 μg/kg/day) for 4 - 5 consecutive days. Leukapheresis should be
started at day 5 and continued until day 6 if needed in order to collect
4 x 10
6
CD34+ cells/kg recipient bodyweight (BW).
Severe chronic neutropenia (SCN)
Congenital neutropenia
The recommended starting dose is 1.2 MU/kg/day (12 μg/kg/day) as a single dose or in divided doses.
Idiopathic or cyclic neutropenia
The recommended starting dose is 0.5 MU/kg/day (5 μg/kg/day) as a single dose or in divided doses.
Dose adjustments
Filgrastim should be administered daily until the neutrophil count has reached and can be maintained
at more than 1.5 x 10
9
/l. When the response has been obtained, the minimal effective dose to maintain
this level should be established. Long-term daily administration is required to maintain an adequate
neutrophil count.
After 1 - 2 weeks of therapy, the initial dose may be doubled or halved depending upon the patient's
response. Subsequently the dose may be individually adjusted every 1 - 2 weeks to maintain the
average neutrophil count between 1.5 x 10
9
/l and 10 x 10
9
/l. A faster schedule of dose escalation may
be considered in patients presenting with severe infections. In clinical studies, 97% of patients who
responded had a complete response at doses of ≤ 2.4 MU/kg/day (24 μg/kg/day). The long-term safety
of filgrastim administration above 2.4 MU/kg/day (24 μg/kg/day) in patients with SCN has not been
established.
HIV infection
Reversal of neutropenia
The recommended starting dose of filgrastim is 0.1 MU/kg/day (1 μg/kg/day) given daily with titration
up to a maximum of 0.4 MU/kg/day (4 μg/kg/day) until a normal neutrophil count is reached and can
be maintained (ANC > 2.0 x 10
9
/l). In clinical studies, > 90% of patients responded at these doses,
achieving reversal of neutropenia in a median of 2 days.
In a small number of patients (< 10%), doses up to 1.0 MU/kg/day (10 μg/kg/day) were required to
achieve reversal of neutropenia.
Maintenance of normal neutrophil counts
When reversal of neutropenia has been achieved, the minimal effective dose to maintain a normal
neutrophil count should be established. Initial dose adjustment to alternate day dosing with
30 MU/day (300 μg/day) is recommended. Further dose adjustment may be necessary, as determined
by the patient's ANC, to maintain the neutrophil count at > 2.0 x 10
9
/l. In clinical studies, dosing with
30 MU/day (300 μg/day) on 1 - 7 days per week was required to maintain the ANC > 2.0 x 10
9
/l, with
the median dose frequency being 3 days per week. Long-term administration may be required to
maintain the ANC > 2.0 x 10
9
/l.
Special populations
Patients with renal/hepatic impairment
Studies of filgrastim in patients with severe impairment of renal or hepatic function demonstrate that it
exhibits a similar pharmacokinetic and pharmacodynamic profile to that seen in normal individuals.
Dose adjustment is not required in these circumstances.
4
Paediatric patients in the SCN and cancer settings
In clinical studies 65% of patients treated for SCN were younger than 18 years. For this age-group,
which mostly includes patients with congenital neutropenia, efficacy was proven. There were no
differences in the safety profiles for paediatric patients treated for SCN in comparison to adults.
Data from clinical studies in paediatric patients indicate that the safety and efficacy of filgrastim are
similar in both adults and children receiving cytotoxic chemotherapy.
The dosage recommendations in paediatric patients are the same as those in adults receiving
myelosuppressive cytotoxic chemotherapy.
Elderly patients
In clinical investigations with filgrastim a small number of elderly patients was included. No specific
studies have been performed for this patient population. Therefore, specific posology
recommendations for these patients cannot be made.
Method of administration
Established cytotoxic chemotherapy
Filgrastim may be administered as a daily subcutaneous injection or alternatively as a daily
intravenous infusion over 30 minutes. For further instructions on dilution with glucose 50 mg/ml (5%)
solution prior to infusion see section 6.6. The subcutaneous route is preferred in most cases. There is
some evidence from a study of single dose administration that intravenous dosing may shorten the
duration of effect. The clinical relevance of this finding to multiple dose administration is not clear.
The choice of route should depend on the individual clinical circumstance. In randomised clinical
studies, a subcutaneous dose of 23 MU/m
2
/day (230 μg/m
2
/day) or rather
0.4 - 0.84 MU/kg/day (4 - 8.4 μg/kg/day) was used.
Patients treated with myeloablative therapy followed by bone marrow transplantation
Filgrastim is administered as an intravenous short-term infusion over 30 minutes or as a subcutaneous
or intravenous continuous infusion over 24 hours, in each case after dilution in 20 ml of glucose
50 mg/ml (5%) solution. For further instructions on dilution with glucose 50 mg/ml (5%) solution
prior to infusion see section 6.6.
Mobilisation of PBPC
Subcutaneous injection.
For the mobilisation of PBPC in patients undergoing myelosuppressive or myeloablative therapy
followed by autologous PBPC transplantation the recommended dose of filgrastim may also be
administered as a 24 hour subcutaneous continuous infusion. For infusions filgrastim should be diluted
in 20 ml of glucose 50 mg/ml (5%) solution. For further instructions on dilution with glucose
50 mg/ml (5%) solution prior to infusion see section 6.6.
SCN/HIV infection
Subcutaneous injection.
Hypersensitivity to the active substance or to any of the excipients.
Special warnings
Filgrastim should not be used to increase the dose of cytotoxic chemotherapy beyond established
posology regimens (see below).
5
Filgrastim should not be administered to patients with severe congenital neutropenia (Kostmann's
syndrome) with abnormal cytogenetics (see below).
Established cytotoxic chemotherapy
Malignant cell growth
Since investigations showed that G-CSF can promote growth of myeloid cells
in vitro
the following
warnings should be considered.
The safety and efficacy of filgrastim administration in patients with myelodysplastic syndrome, or
chronic myelogenous leukaemia have not been established. Therefore filgrastim is not indicated for
use in these conditions. Particular care should be taken to distinguish the diagnosis of blast
transformation of chronic myeloid leukaemia from acute myeloid leukaemia.
In view of limited safety and efficacy data in patients with secondary AML, filgrastim should be
administered with caution.
The safety and efficacy of filgrastim administration in
de novo
AML patients aged < 55 years with
good cytogenetics [t(8;21), t(15;17), and inv(16)] have not been established.
Leucocytosis
White blood cell counts of 100 x 10
9
/l or greater have been observed in less than 5% of patients
receiving filgrastim at doses above 0.3 MU/kg/day (3 μg/kg/day). No undesirable effects directly
attributable to this degree of leucocytosis have been reported. However, in view of the potential risks
associated with severe leucocytosis, a white blood cell count should be performed at regular intervals
during filgrastim therapy. If leukocyte counts exceed 50 x 10
9
/l after the expected nadir, filgrastim
should be discontinued immediately. However, during the period of administration of filgrastim for
PBPC mobilisation, it should be discontinued or its dose should be reduced if the leukocyte counts rise
to > 70 x 10
9
/l.
Risks associated with increased doses of chemotherapy
Special caution should be used when treating patients with high-dose chemotherapy because improved
tumour outcome has not been demonstrated and intensified doses of chemotherapeutic agents may lead
to increased toxicities including cardiac, pulmonary, neurologic, and dermatologic effects (please refer
to the Summary of Product Characteristics of the specific chemotherapy agents used).
Treatment with filgrastim alone does not preclude thrombocytopenia and anaemia due to
myelosuppressive chemotherapy. Because of the potential of receiving higher doses of chemotherapy
(e.g. full doses on the prescribed schedule) the patient may be at greater risk of thrombocytopenia and
anaemia. Regular monitoring of platelet count and haematocrit is recommended. Special care should
be taken when administering single or combination chemotherapeutic agents which are known to
cause severe thrombocytopenia.
The use of filgrastim-mobilised PBPCs has been shown to reduce the depth and duration of
thrombocytopenia following myelosuppressive or myeloablative chemotherapy.
Other special precautions
The effects of filgrastim in patients with substantially reduced myeloid progenitors have not been
studied. Filgrastim acts primarily on neutrophil precursors to exert its effect in elevating neutrophil
counts. Therefore, in patients with reduced precursors, neutrophil response may be diminished (such
as those treated with extensive radiotherapy or chemotherapy, or those with bone marrow infiltration
by tumour).
There have been reports of Graft versus Host Disease (GvHD) and fatalities in patients receiving
G-CSF after allogeneic bone marrow transplantation (see section 5.1).
6
Mobilisation of PBPC
Prior exposure to cytotoxic agents
Patients who have undergone very extensive prior myelosuppressive therapy, followed by
administration of filgrastim for mobilisation of PBPCs, may not show sufficient mobilisation of these
blood cells to achieve the recommended minimum yield (≥ 2.0 x 10
6
CD34+ cells/kg) or acceleration
of platelet recovery to the same degree.
Some cytotoxic agents exhibit particular toxicities to the haematopoietic progenitor pool and may
adversely affect progenitor mobilisation. Agents such as melphalan, carmustine (BCNU) and
carboplatin, when administered over prolonged periods prior to attempts at progenitor mobilisation
may reduce progenitor yield. However, the administration of melphalan, carboplatin or BCNU
together with filgrastim has been shown to be effective for progenitor mobilisation. When a PBPC
transplantation is envisaged it is advisable to plan the stem cell mobilisation procedure early in the
treatment course of the patient. Particular attention should be paid to the number of progenitors
mobilised in such patients before the administration of high-dose chemotherapy. If yields are
inadequate, as measured by the criteria above, alternative forms of treatment not requiring progenitor
support should be considered.
Assessment of progenitor cell yields
In assessing the number of progenitor cells harvested in patients treated with filgrastim, particular
attention should be paid to the method of quantitation. The results of flow cytometric analysis of
CD34+ cell numbers vary depending on the precise methodology used and therefore,
recommendations of numbers based on studies in other laboratories need to be interpreted with
caution.
Statistical analysis of the relationship between the number of CD34+ cells re-infused and the rate of
platelet recovery after high-dose chemotherapy indicates a complex but continuous relationship. The
recommendation of a minimum yield of ≥ 2.0 x 10
6
CD34+ cells/kg is based on published experience
resulting in adequate haematologic reconstitution. Yields in excess of this minimum yield appear to
correlate with more rapid recovery, those below with slower recovery.
Normal donors prior to allogeneic PBPC transplantation
Mobilisation of PBPC does not provide a direct clinical benefit to normal donors and should only be
considered for the purposes of allogeneic stem cell transplantation.
PBPC mobilisation should be considered only in donors who meet normal clinical and laboratory
eligibility criteria for stem cell donation with special attention to haematological values and infectious
disease.
The safety and efficacy of filgrastim have not been assessed in normal donors < 16 years or
> 60 years.
Transient thrombocytopenia (platelets < 100 x 10
9
/l) following filgrastim administration and
leukapheresis was observed in 35% of subjects studied. Among these, two cases of platelets
< 50 x 10
9
/l were reported and attributed to the leukapheresis procedure. If more than one
leukapheresis is required, particular attention should be paid to donors with platelets < 100 x 10
9
/l
prior to leukapheresis; in general apheresis should not be performed if platelets < 75 x 10
9
/l.
Leukapheresis should not be performed in donors who are anticoagulated or who have known defects
in haemostasis.
Filgrastim administration should be discontinued or its posology should be reduced if the leukocyte
counts rise to > 70 x 10
9
/l.
Donors who receive G-CSFs for PBPC mobilisation should be monitored until haematological indices
return to normal.
7
Transient cytogenetic modifications have been observed in normal donors following G-CSF use. The
significance of these changes is unknown.
Long-term safety follow-up of donors is ongoing. Nevertheless, a risk of promotion of a malignant
myeloid clone can not be excluded. It is recommended that the apheresis centre perform a systematic
record and tracking of the stem cell donors for at least 10 years to ensure monitoring of long-term
safety.
Common but generally asymptomatic cases of splenomegaly and very rare cases of splenic rupture
have been reported in healthy donors and patients following administration of G-CSFs. Some cases of
splenic rupture were fatal. Therefore, spleen size should be carefully monitored (e.g. clinical
examination, ultrasound). A diagnosis of splenic rupture should be considered in donors and/or
patients reporting left upper abdominal pain or shoulder tip pain.
In normal donors, pulmonary adverse events (haemoptysis, pulmonary haemorrhage, pulmonary
infiltrates, dyspnoea and hypoxia) have been reported very rarely in post marketing experience. In
case of suspected or confirmed pulmonary adverse events, discontinuation of treatment with filgrastim
should be considered and appropriate medical care given.
Recipients of allogeneic PBPCs mobilised with filgrastim
Current data indicate that immunological interactions between the allogeneic PBPC graft and the
recipient may be associated with an increased risk of acute and chronic GvHD when compared with
bone marrow transplantation.
SCN
Blood cell counts
Platelet counts should be monitored closely, especially during the first few weeks of filgrastim
therapy. Consideration should be given to intermittent cessation or decreasing the dose of filgrastim in
patients who develop thrombocytopenia, i.e. platelets consistently < 100,000/mm
3
.
Other blood cell changes occur, including anaemia and transient increases in myeloid progenitors,
which require close monitoring of cell counts.
Transformation to leukaemia or myelodysplastic syndrome
Special care should be taken in the diagnosis of SCNs to distinguish them from other haematopoietic
disorders such as aplastic anaemia, myelodysplasia, and myeloid leukaemia. Complete blood cell
counts with differential and platelet counts, and an evaluation of bone marrow morphology and
karyotype should be performed prior to treatment.
There was a low frequency (approximately 3%) of myelodysplastic syndromes (MDS) or leukaemia in
clinical study patients with SCN treated with filgrastim. This observation has only been made in
patients with congenital neutropenia. MDS and leukaemias are natural complications of the disease
and are of uncertain relation to filgrastim therapy. A subset of approximately 12% of patients who had
normal cytogenetic evaluations at baseline were subsequently found to have abnormalities, including
monosomy 7, on routine repeat evaluation. If patients with SCN develop abnormal cytogenetics, the
risks and benefits of continuing filgrastim should be carefully weighed; Filgrastim should be
discontinued if MDS or leukaemia occurs. It is currently unclear whether long-term treatment of
patients with SCN will predispose patients to cytogenetic abnormalities, MDS or leukaemic
transformation. It is recommended to perform morphologic and cytogenetic bone marrow
examinations in patients at regular intervals (approximately every 12 months).
Other special precautions
Causes of transient neutropenia, such as viral infections should be excluded.
Splenic enlargement is a direct effect of treatment with filgrastim. 31% of patients in studies were
documented as having palpable splenomegaly. Increases in volume, measured radiographically,
occurred early during Filgrastim therapy and tended to plateau. Dose reductions were noted to slow or
8
stop the progression of splenic enlargement, and in 3% of patients a splenectomy was required. Spleen
size should be evaluated regularly. Abdominal palpation should be sufficient to detect abnormal
increases in splenic volume.
Haematuria/proteinuria occurred in a small number of patients. Regular urine analyses should be
performed to monitor this event.
The safety and efficacy in neonates and patients with autoimmune neutropenia have not been
established.
HIV infection
Blood cell counts
ANC should be monitored closely, especially during the first few weeks of filgrastim therapy. Some
patients may respond very rapidly and with a considerable increase in neutrophil count to the initial
dose of filgrastim. It is recommended that the ANC is measured daily for the first 2 - 3 days of
filgrastim administration. Thereafter, it is recommended that the ANC is measured at least twice per
week for the first 2 weeks and subsequently once per week or once every other week during
maintenance therapy. During intermittent dosing with 30 MU/day (300 μg/day) of filgrastim, there can
be wide fluctuations in the patient's ANC over time. In order to determine a patient's trough or nadir
ANC, it is recommended that blood samples are taken for ANC measurement immediately prior to any
scheduled dosing with filgrastim.
Risk associated with increased doses of myelosuppressive medicinal products
Treatment with filgrastim alone does not preclude thrombocytopenia and anaemia due to
myelosuppressive treatments. As a result of the potential to receive higher doses or a greater number
of these medicinal products with filgrastim therapy, the patient may be at higher risk of developing
thrombocytopenia and anaemia. Regular monitoring of blood counts is recommended (see above).
Infections and malignancies causing myelosuppression
Neutropenia may be due to bone marrow-infiltrating opportunistic infections such as Mycobacterium
avium complex or malignancies such as lymphoma. In patients with known bone marrow-infiltrating
infections or malignancy, consider appropriate therapy for treatment of the underlying condition in
addition to administration of filgrastim for treatment of neutropenia. The effects of filgrastim on
neutropenia due to bone marrow-infiltrating infection or malignancy have not been well established.
Other special precautions
Rare pulmonary adverse reactions, in particular interstitial pneumonia, have been reported after
G-CSF administration (see section 4.8). Patients with a recent history of pulmonary infiltrates or
pneumonia may be at higher risk. The onset of pulmonary signs, such as cough, fever and dyspnoea in
association with radiological signs of pulmonary infiltrates and deterioration in pulmonary function
may be preliminary signs of Adult Respiratory Distress Syndrome (ARDS). Filgrastim should be
discontinued and appropriate treatment given in these cases.
Monitoring of bone density may be indicated in patients with underlying osteoporotic bone diseases
who undergo continuous therapy with filgrastim for more than 6 months.
Sickle cells crises, in some cases fatal, have been reported with the use of filgrastim in subjects with
sickle cell disease. Physicians should exercise caution when considering the use of filgrastim in
patients with sickle cell disease, and only after careful evaluation of the potential risks and benefits.
Increased haematopoietic activity of the bone marrow in response to growth factor therapy has been
associated with transient positive bone-imaging findings. This should be considered when interpreting
bone imaging results.
9
Excipients
Zarzio contains sorbitol. Patients with rare hereditary problems of fructose intolerance should not use
this medicinal product.
The safety and efficacy of filgrastim given on the same day as myelosuppressive cytotoxic
chemotherapy have not been definitively established. In view of the sensitivity of rapidly dividing
myeloid cells to myelosuppressive cytotoxic chemotherapy, the use of filgrastim is not recommended
in the period from 24 hours before to 24 hours after chemotherapy. Preliminary evidence from a small
number of patients treated concomitantly with filgrastim and 5-fluorouracil indicates that the severity
of neutropenia may be exacerbated.
Possible interactions with other haematopoietic growth factors and cytokines have not yet been
investigated in clinical studies.
Since lithium promotes the release of neutrophils, lithium is likely to potentiate the effect of filgrastim.
Although this interaction has not been formally investigated, there is no evidence that such an
interaction is harmful.
There are no adequate data from the use of filgrastim in pregnant women. There are reports in the
literature where the transplacental passage of filgrastim in pregnant women has been demonstrated.
There is no evidence from studies in rats and rabbits that filgrastim is teratogenic. An increased
incidence of embryo-loss has been observed in rabbits, but no malformation has been seen.
In pregnancy, the possible risk of filgrastim use to the foetus must be weighed against the expected
therapeutic benefit.
It is not known whether filgrastim is excreted in human milk, therefore it is not recommended for use
in breast-feeding women.
Filgrastim has no influence on the ability to drive and use machines.
The most commonly reported adverse reactions to filgrastim are mild to moderate musculoskeletal
pain occurring in more than 10% of patients. Musculoskeletal pain is usually controlled with standard
analgesics.
Adverse reactions listed below are classified according to frequency and system organ class.
Frequency groupings are defined according to the following convention: Very common (≥ 1/10),
common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very
rare (< 1/10,000), not known (cannot be estimated from the available data).
10
Table 1. Adverse reactions in clinical trials in cancer patients
Immune system disorders
Very rare:
Allergic-type reactions*, including anaphylaxis, skin rash, urticaria,
angioedema, dyspnoea and hypotension
Vascular disorders
Uncommon: Hypotension (transient)
Rare: Vascular disorders* including venoocclusive disease and fluid volume
disturbances
Respiratory, thoracic and mediastinal disorders
Very rare: Pulmonary oedema*, interstitial pneumonia*, pulmonary infiltrates*
Skin and subcutaneous tissue disorders
Very rare: Sweet's Syndrome*, cutaneous vasculitis
Musculoskeletal, connective tissue and bone disorders
Very common: Musculoskeletal pain (mild or moderate)
Common: Musculoskeletal pain (severe)
Very rare: Rheumatoid arthritis exacerbation
Renal and urinary disorders
Very rare:
Micturition disorders (predominantly dysuria)
Investigations
Very common: Blood alkaline phosphatase, blood lactate dehydrogenase (LDH),
gamma-glutamyltransferase (GGT) and blood uric acid increased
(reversible, dose-dependent, mild or moderate)
* see below
Table 2. Adverse reactions in clinical trials in normal donors undergoing PBPC mobilisation
Immune system disorders
Uncommon: Severe allergic reaction: anaphylaxis, angioedema, urticaria, rash
Blood and the lymphatic system disorders
Very common: Leucocytosis (WBC > 50 x 10
9
/l), thrombocytopenia (platelets
< 100 x 10
9
/l; transient)
Common: Splenomegaly (generally asymptomatic, also in patients)
Uncommon: Spleen disorder
Very rare: Splenic rupture (also in patients)
Nervous system disorders
Very common: Headache
Respiratory, thoracic and mediastinal disorders
Very rare
:
Haemoptysis*, pulmonary haemorrhage*, pulmonary infiltrates*,
dyspnoea*, hypoxia*
Musculoskeletal, connective tissue and bone disorders
Very common: Musculoskeletal pain (mild to moderate, transient)
Uncommon: Rheumatoid arthritis and arthritic symptoms exacerbation
Investigations
Common: Blood alkaline phosphatase and LDH increased (transient, minor)
Uncommon: Aspartate aminotransferase (AST) and blood uric acid increased
(transient, minor)
* see below
11














Table 3. Adverse reactions in clinical trials in SCN patients
Blood and the lymphatic system disorders
Very common: Anaemia, splenomegaly (may be progressive in a minority of cases)
Common: Thrombocytopenia
Uncommon: Spleen disorder
Nervous system disorders
Common: Headache
Respiratory, thoracic and mediastinal disorders
Very common: Epistaxis
Gastrointestinal disorders
Common: Diarrhoea
Hepato-biliary disorders
Common: Hepatomegaly
Skin and subcutaneous tissue disorders
Common: Cutaneous vasculitis (during long term use), alopecia, rash
Musculoskeletal, connective tissue and bone disorders
Very common: General musculoskeletal pain, bone pain
Common: Osteoporosis, arthralgia
Renal and urinary disorders
Uncommon: Haematuria, proteinuria
General disorders and administration site conditions
Common:
Investigations
Very common: Blood alkaline phosphatase, LDH and blood uric acid increased
(transient), blood glucose decreased (transient, moderate)
Table 4. Adverse reactions in clinical trials in HIV Patients
Blood and lymphatic system disorders
Common: Spleen disorder, splenomegaly*
Musculoskeletal, connective tissue and bone disorders
Very common: Musculoskeletal pain (mild to moderate)
* see below
In randomised, placebo-controlled clinical studies, filgrastim did not increase the incidence of
frequency in patients treated with filgrastim/chemotherapy and placebo/chemotherapy included nausea
and vomiting, alopecia, diarrhoea, fatigue, anorexia, mucositis, headache, cough, skin rash, chest pain,
generalised weakness, sore throat, constipation and unspecified pain.
Allergic reactions occurred on initial or subsequent treatment in patients receiving filgrastim. Overall,
reports were more common after intravenous administration. In some cases, symptoms have recurred
with rechallenge, suggesting a causal relationship. Filgrastim should be permanently discontinued in
patients who experience a serious allergic reaction.
Vascular disorders have been reported in patients undergoing high dose chemotherapy followed by
autologous bone marrow transplantation. The causal association with filgrastim has not been
established.
Pulmonary adverse reactions have in some cases been reported with an outcome of respiratory failure
or adult respiratory distress syndrome (ARDS), which may be fatal. In normal donors, pulmonary
adverse events (haemoptysis, pulmonary haemorrhage, pulmonary infiltrates, dyspnoea and hypoxia)
have been reported very rarely in post marketing experience (see section 4.4).
The occurrence of Sweet's syndrome (acute febrile neutrophilic dermatosis) has been reported
occasionally in cancer patients. However, since a significant percentage of these patients were
12
Injection site pain













suffering from leukaemia, a condition known to be associated with Sweet's syndrome, a causal
relationship with filgrastim has not been established.
Isolated cases of sickle cells crises have been reported in patients with sickle cell disease (see section
4.4). The frequency is not known.
In all cases of splenic enlargement in HIV patients this was mild or moderate on physical examination
and the clinical course was benign; no patients had a diagnosis of hypersplenism and no patients
underwent splenectomy. As splenic enlargement is a common finding in patients with HIV infection
and is present to varying degrees in most patients with AIDS, the relationship to filgrastim treatment is
unclear.
Immunogenicity
In four clinical studies none of the healthy volunteers or cancer patients developed anti-rhG-CSF
antibodies (neither binding nor neutralising) upon treatment with Zarzio.
The effects of filgrastim overdose have not been established.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Colony stimulating factors, ATC Code: L03AA02
Human G-CSF is a glycoprotein which regulates the production and release of functional neutrophils
from the bone marrow. Zarzio containing r-metHuG-CSF (filgrastim) causes marked increases in
peripheral blood neutrophil counts within 24 hours, with minor increases in monocytes. In some SCN
patients filgrastim can also induce a minor increase in the number of circulating eosinophils and
basophils relative to baseline; some of these patients may present with eosinophilia or basophilia
already prior to treatment. Elevations of neutrophil counts are dose-dependent at recommended doses.
Neutrophils produced in response to filgrastim show normal or enhanced function as demonstrated by
tests of chemotactic and phagocytic function. Following termination of filgrastim therapy, circulating
neutrophil counts decrease by 50% within 1 - 2 days, and to normal levels within 1 - 7 days. As with
other haematopoietic growth factors, G-CSF has shown
in vitro
stimulating properties on human
endothelial cells, which have specific receptors for G-CSF. Accordingly, G-CSF has been shown to
induce endothelial cell functions related to angiogenesis. Additionally, G-CSF has been shown to
increase neutrophil migration across the vascular endothelium.
Use of filgrastim in patients undergoing cytotoxic chemotherapy
leads to significant reductions in the
incidence, severity and duration of neutropenia and febrile neutropenia. Treatment with filgrastim
significantly reduces the duration of febrile neutropenia, antibiotic use and hospitalisation after
induction chemotherapy for acute myelogenous leukaemia or myeloablative therapy followed by bone
marrow transplantation. The incidence of fever and documented infections were not reduced in either
setting. The duration of fever was not reduced in patients undergoing myeloablative therapy followed
by bone marrow transplantation.
Use of filgrastim, either alone, or after chemotherapy,
mobilises haematopoietic progenitor cells into
peripheral blood. These autologous PBPCs may be harvested and infused after high-dose cytotoxic
therapy, either in place of, or in addition to bone marrow transplantation. Infusion of PBPCs
accelerates haematopoietic recovery reducing the duration of risk for haemorrhagic complications and
the need for platelet transfusions.
One retrospective European study evaluating the use of G-CSF after allogeneic bone marrow
transplantation in patients with acute leukaemias suggested an increase in the risk of GvHD, treatment
13
related mortality (TRM) and mortality when G-CSF was administered. In a separate retrospective
international study in patients with acute and chronic myelogenous leukaemias, no effect on the risk of
GvHD, TRM and mortality was seen . A meta-analysis of allogeneic transplant studies, including the
results of nine prospective randomized trials, 8 retrospective studies and 1 case-controlled study, did
not detect an effect on the risks of acute GvHD, chronic GvHD or early treatment-related mortality.
Relative Risk (95% CI) of GvHD and TRM
Following treatment with G-CSF after bone marrow transplantation
Publication
Period of
Study
N
Acute Grade
II - IV GvHD
Chronic
GvHD
TRM
Meta-Analysis
(2003)
1986 - 2001
a
1198
1.08
(0.87, 1.33)
1.02
(0.82, 1.26)
0.70
(0.38, 1.31)
European
Retrospective
Study (2004)
1992 - 2002
b
1789
1.33
(1.08, 1.64)
1.29
(1.02, 1.61)
1.73
(1.30, 2.32)
1.26
(0.95, 1.67)
a
Analysis includes studies involving BM transplant during this period; some studies used GM-CSF
b
Analysis includes patients receiving BM transplant during this period
1995 - 2000
b
2110
1.11
(0.86, 1.42)
1.10
(0.86, 1.39)
Use of filgrastim for the mobilisation of PBPCs in normal donors prior to allogeneic PBPC
transplantation
In normal donors, a 1 MU/kg/day (10 μg/kg/day) dose administered subcutaneously for
4 - 5 consecutive days allows a collection of ≥ 4 x 10
6
CD34+ cells/kg recipient BW in the majority of
the donors after two leukaphereses.
Recipients of allogeneic PBPCs mobilised with filgrastim experienced significantly more rapid
haematological recovery, leading to a significant decrease in time to unsupported platelet recovery
when compared with allogeneic bone marrow transplantation.
Use of filgrastim in children or adults with SCN
(severe congenital, cyclic, and idiopathic neutropenia)
induces a sustained increase in ACNs in peripheral blood and a reduction of infection and related
events.
Use of filgrastim in patients with HIV infection
maintains normal neutrophil counts to allow scheduled
dosing of antiviral and/or other myelosuppressive treatments. There is no evidence that patients with
HIV infection treated with filgrastim show an increase in HIV replication.
5.2 Pharmacokinetic properties
Randomised, double-blind, single and multiple dose, crossover studies in 146 healthy volunteers
showed that the pharmacokinetic profile of Zarzio was comparable to that of the reference product
after subcutaneous and intravenous administration.
Absorption
A single subcutaneous dose of 0.5 MU/kg (5 µg/kg) resulted in maximum serum concentrations after a
t
max
of 4.5 ± 0.9 hours (mean ± SD).
Distribution
The volume of distribution in blood is approximately 150 ml/kg. Following subcutaneous
administration of recommended doses, serum concentrations were maintained above 10 ng/ml for
8 - 16 hours. There is a positive linear correlation between the dose and the serum concentration of
filgrastim, whether administered intravenously or subcutaneously.
14
International
Retrospective
Study (2006)












Elimination
The elimination of filgrastim is non-linear with respect to dose, the serum clearance decreases with
increasing dose. Filgrastim appears to be mainly eliminated by neutrophil mediated clearance, which
becomes saturated at higher doses. However, the serum clearance increases with repeated dosing while
the neutrophil count increases. The median serum elimination half-life (t
1/2
) of filgrastim after single
subcutaneous doses ranged from 2.7 hours (1.0 MU/kg, 10 µg/kg) to 5.7 hours (0.25 MU/kg,
2.5 µg/kg) and was prolonged after 7 days of dosing to 8.5 - 14 hours, respectively.
Continuous infusion with filgrastim over a period of up to 28 days, in patients recovering from
autologous bone-marrow transplantation, resulted in no evidence of drug accumulation and
comparable elimination half-lives.
5.3 Preclinical safety data
There are no preclinical data of relevance to the prescriber which are additional to that already
included in other sections of the Summary of Product Characteristics.
6.
6.1 List of excipients
Glutamic acid
Sorbitol (E420)
Polysorbate 80
Water for injections
6.2 Incompatibilities
Zarzio must not be diluted with sodium chloride solutions.
This medicinal product must not be mixed with other medicinal products except those mentioned in
section 6.6.
Diluted filgrastim may be adsorbed to glass and plastic materials, unless it is diluted in
glucose 50 mg/ml (5%) solution (see section 6.6).
6.3 Shelf life
30 months.
After dilution: Chemical and physical in-use stability of the diluted solution for infusion has been
demonstrated for 24 hours at 2°C to 8°C. From a microbiological point of view, the product should be
used immediately. If not used immediately, in-use storage times and conditions prior to use are the
responsibility of the user and would normally not be longer than 24 hours at 2°C to 8°C, unless
dilution has taken place in controlled and validated aseptic conditions.
6.4 Special precautions for storage
Store in a refrigerator (2°C - 8°C).
Keep the pre-filled syringe in the outer carton in order to protect from light.
Within its shelf-life and for the purpose of ambulatory use, the patient may remove the product from
the refrigerator and store it at room temperature (not above 25°C) for one single period of up to 72
hours. At the end of this period, the product should not be put back in the refrigerator and should be
disposed of.
15
For storage conditions of the diluted medicinal product, see section 6.3.
6.5 Nature and contents of container
Pre-filled syringe (type I glass) with injection needle (stainless steel), with or without a needle safety
guard, containing 0.5 ml solution.
Pack sizes of 1, 3, 5 or 10 pre-filled syringes.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
The solution should be visually inspected prior to use. Only clear solutions without particles should be
used. Accidental exposure to freezing temperatures does not adversely affect the stability of Zarzio.
Zarzio contains no preservative: In view of the possible risk of microbial contamination, Zarzio
syringes are for single use only.
Dilution prior to administration (optional)
If required, Zarzio may be diluted in glucose 50 mg/ml (5%) solution.
Dilution to a final concentration < 0.2 MU/ml (2 μg/ml) is not recommended at any time.
For patients treated with filgrastim diluted to concentrations < 1.5 MU/ml (15 μg/ml), human serum
albumin (HSA) should be added to a final concentration of 2 mg/ml.
Example: In a final volume of 20 ml, total doses of filgrastim less than 30 MU (300 μg) should be
given with 0.2 ml of human serum albumin 200 mg/ml (20%) solution Ph. Eur. added.
When diluted in glucose 50 mg/ml (5%) solution, filgrastim is compatible with glass and a variety of
plastics including polyvinylchloride, polyolefin (a copolymer of polypropylene and polyethylene) and
polypropylene.
Using the pre-filled syringe with a needle safety guard
The needle safety guard covers the needle after injection to prevent needle stick injury. This does not
affect normal operation of the syringe. Depress the plunger slowly and evenly until the entire dose has
been given and the plunger cannot be depressed any further. While maintaining pressure on the
plunger, remove the syringe from the patient. The needle safety guard will cover the needle when
releasing the plunger.
Using the pre-filled syringe without a needle safety guard
Administer the dose as per standard protocol.
Disposal
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Sandoz GmbH
Biochemiestrasse 10
A-6250 Kundl
Austria
16
8.
EU/1/08/495/001
EU/1/08/495/002
EU/1/08/495/003
EU/1/08/495/004
EU/1/08/495/009
EU/1/08/495/010
EU/1/08/495/011
EU/1/08/495/012
9.
06/02/2009
Detailed information on this medicinal product is available on the website of the European Medicines
Agency (EMA)
http://www.ema.europa.eu/
.
17
1.
Zarzio 48 MU/0.5 ml solution for injection or infusion in pre-filled syringe
2.
Each ml of solution contains 96 million units (MU) [equivalent to 960 micrograms (μg)] filgrastim*.
Each pre-filled syringe contains 48 MU (equivalent to 480 μg) filgrastim in 0.5 ml.
* recombinant methionylated human granulocyte-colony stimulating factor (G-CSF) produced in
E. coli
by recombinant DNA technology.
Excipient: Each ml of solution contains 50 mg sorbitol (E420).
For a full list of excipients, see section 6.1.
3.
Solution for injection or infusion in pre-filled syringe
Clear, colourless to slightly yellowish solution.
4.
-
Reduction in the duration of neutropenia and the incidence of febrile neutropenia in patients
treated with established cytotoxic chemotherapy for malignancy (with the exception of chronic
myeloid leukaemia and myelodysplastic syndromes) and reduction in the duration of
neutropenia in patients undergoing myeloablative therapy followed by bone marrow
transplantation considered to be at increased risk of prolonged severe neutropenia.
The safety and efficacy of filgrastim are similar in adults and children receiving cytotoxic
chemotherapy.
-
Mobilisation of peripheral blood progenitor cells (PBPC).
-
In children and adults with severe congenital, cyclic, or idiopathic neutropenia with an absolute
neutrophil count (ANC) of ≤ 0.5 x 10
9
/l, and a history of severe or recurrent infections, long
term administration of filgrastim is indicated to increase neutrophil counts and to reduce the
incidence and duration of infection-related events.
-
Treatment of persistent neutropenia (ANC ≤ 1.0 x 10
9
/l) in patients with advanced HIV
infection, in order to reduce the risk of bacterial infections when other therapeutic options are
inappropriate.
Filgrastim therapy should only be given in collaboration with an oncology centre which has
experience in granulocyte-colony stimulating factor (G-CSF) treatment and haematology and has the
necessary diagnostic facilities.
18
The mobilisation and apheresis procedures should be performed in collaboration with an
oncology-haematology centre with acceptable experience in this field and where the monitoring of
haematopoietic progenitor cells can be correctly performed.
Zarzio is available in strengths of 30 MU/0.5 ml and 48 MU/0.5 ml.
Established cytotoxic chemotherapy
The recommended dose of filgrastim is 0.5 MU/kg/day (5 μg/kg/day). The first dose of filgrastim
should not be administered less than 24 hours following cytotoxic chemotherapy.
Daily dosing with filgrastim should continue until the expected neutrophil nadir is passed and the
neutrophil count has recovered to the normal range. Following established chemotherapy for solid
tumours, lymphomas, and lymphoid leukaemias, it is expected that the duration of treatment required
to fulfil these criteria will be up to 14 days. Following induction and consolidation treatment for acute
myeloid leukaemia the duration of treatment may be substantially longer (up to 38 days) depending on
the type, dose and schedule of cytotoxic chemotherapy used.
In patients receiving cytotoxic chemotherapy, a transient increase in neutrophil counts is typically seen
1 - 2 days after initiation of filgrastim therapy. However, for a sustained therapeutic response,
filgrastim therapy should not be discontinued before the expected nadir has passed and the neutrophil
count has recovered to the normal range. Premature discontinuation of filgrastim therapy, prior to the
time of the expected neutrophil nadir, is not recommended.
Patients treated with myeloablative therapy followed by bone marrow transplantation
The recommended starting dose of filgrastim is 1.0 MU/kg/day (10 μg/kg/day). The first dose of
filgrastim should not be administered less than 24 hours following cytotoxic chemotherapy and within
24 hours of bone marrow infusion.
Dose adjustments:
Once the neutrophil nadir has been passed, the daily dose of filgrastim should be
titrated against the neutrophil response as follows:
Absolute neutrophil count
Filgrastim dose adjustment
ANC > 1.0 x 10
9
/l for 3 consecutive days
Reduce to 0.5 MU/kg/day (5 μg/kg/day)
Then, if ANC remains > 1.0 x 10
9
/l for
3 more consecutive days
Discontinue filgrastim
If the ANC decreases to < 1.0 x 10
9
/l during the treatment period, the dose of filgrastim
should be re-escalated according to the above steps
Mobilisation of PBPC
Patients undergoing myelosuppressive or myeloablative therapy followed by autologous PBPC
transplantation
The recommended dose of filgrastim for PBPC mobilisation when used alone is
1.0 MU/kg/day (10 μg/kg/day) for 5 - 7 consecutive days. Timing of leukapheresis:
1 or 2 leukaphereses on days 5 and 6 are often sufficient. In other circumstances, additional
leukaphereses may be necessary. Filgrastim dosing should be maintained until the last leukapheresis.
The recommended dose of filgrastim for PBPC mobilisation after myelosuppressive chemotherapy is
0.5 MU/kg/day (5 μg/kg/day) given daily from the first day after completion of chemotherapy until the
expected neutrophil nadir is passed and the neutrophil count has recovered to the normal range.
Leukapheresis should be performed during the period when the ANC rises from < 0.5 x 10
9
/l to
> 5.0 x 10
9
/l. For patients who have not had extensive chemotherapy, one leukapheresis is often
sufficient. In other circumstances, additional leukaphereses are recommended.
There are no prospectively randomised comparisons of the two recommended mobilisation methods
(filgrastim alone or in combination with myelosuppressive chemotherapy) within the same patient
population. The degree of variation between individual patients and between laboratory assays of
19







CD34
+
cells means that direct comparison between different studies is difficult. It is therefore difficult
to recommend an optimum method. The choice of mobilisation method should be considered in
relation to the overall objectives of treatment for an individual patient.
Normal donors prior to allogeneic PBPC transplantation
For PBPC mobilisation in normal donors prior to allogeneic PBPC transplantation, filgrastim should
be administered at 1.0 MU/kg/day (10 μg/kg/day) for 4 - 5 consecutive days. Leukapheresis should be
started at day 5 and continued until day 6 if needed in order to collect
4 x 10
6
CD34+ cells/kg recipient bodyweight (BW).
Severe chronic neutropenia (SCN)
Congenital neutropenia
The recommended starting dose is 1.2 MU/kg/day (12 μg/kg/day) as a single dose or in divided doses.
Idiopathic or cyclic neutropenia
The recommended starting dose is 0.5 MU/kg/day (5 μg/kg/day) as a single dose or in divided doses.
Dose adjustments
Filgrastim should be administered daily until the neutrophil count has reached and can be maintained
at more than 1.5 x 10
9
/l. When the response has been obtained, the minimal effective dose to maintain
this level should be established. Long-term daily administration is required to maintain an adequate
neutrophil count.
After 1 - 2 weeks of therapy, the initial dose may be doubled or halved depending upon the patient's
response. Subsequently the dose may be individually adjusted every 1 - 2 weeks to maintain the
average neutrophil count between 1.5 x 10
9
/l and 10 x 10
9
/l. A faster schedule of dose escalation may
be considered in patients presenting with severe infections. In clinical studies, 97% of patients who
responded had a complete response at doses of ≤ 2.4 MU/kg/day (24 μg/kg/day). The long-term safety
of filgrastim administration above 2.4 MU/kg/day (24 μg/kg/day) in patients with SCN has not been
established.
HIV infection
Reversal of neutropenia
The recommended starting dose of filgrastim is 0.1 MU/kg/day (1 μg/kg/day) given daily with titration
up to a maximum of 0.4 MU/kg/day (4 μg/kg/day) until a normal neutrophil count is reached and can
be maintained (ANC > 2.0 x 10
9
/l). In clinical studies, > 90% of patients responded at these doses,
achieving reversal of neutropenia in a median of 2 days.
In a small number of patients (< 10%), doses up to 1.0 MU/kg/day (10 μg/kg/day) were required to
achieve reversal of neutropenia.
Maintenance of normal neutrophil counts
When reversal of neutropenia has been achieved, the minimal effective dose to maintain a normal
neutrophil count should be established. Initial dose adjustment to alternate day dosing with
30 MU/day (300 μg/day) is recommended. Further dose adjustment may be necessary, as determined
by the patient's ANC, to maintain the neutrophil count at > 2.0 x 10
9
/l. In clinical studies, dosing with
30 MU/day (300 μg/day) on 1 - 7 days per week was required to maintain the ANC > 2.0 x 10
9
/l, with
the median dose frequency being 3 days per week. Long-term administration may be required to
maintain the ANC > 2.0 x 10
9
/l.
Special populations
Patients with renal/hepatic impairment
Studies of filgrastim in patients with severe impairment of renal or hepatic function demonstrate that it
exhibits a similar pharmacokinetic and pharmacodynamic profile to that seen in normal individuals.
Dose adjustment is not required in these circumstances.
20
Paediatric patients in the SCN and cancer settings
In clinical studies 65% of patients treated for SCN were younger than 18 years. For this age-group,
which mostly includes patients with congenital neutropenia, efficacy was proven. There were no
differences in the safety profiles for paediatric patients treated for SCN in comparison to adults.
Data from clinical studies in paediatric patients indicate that the safety and efficacy of filgrastim are
similar in both adults and children receiving cytotoxic chemotherapy.
The dosage recommendations in paediatric patients are the same as those in adults receiving
myelosuppressive cytotoxic chemotherapy.
Elderly patients
In clinical investigations with filgrastim a small number of elderly patients was included. No specific
studies have been performed for this patient population. Therefore, specific posology
recommendations for these patients cannot be made.
Method of administration
Established cytotoxic chemotherapy
Filgrastim may be administered as a daily subcutaneous injection or alternatively as a daily
intravenous infusion over 30 minutes. For further instructions on dilution with glucose 50 mg/ml (5%)
solution prior to infusion see section 6.6. The subcutaneous route is preferred in most cases. There is
some evidence from a study of single dose administration that intravenous dosing may shorten the
duration of effect. The clinical relevance of this finding to multiple dose administration is not clear.
The choice of route should depend on the individual clinical circumstance. In randomised clinical
studies, a subcutaneous dose of 23 MU/m
2
/day (230 μg/m
2
/day) or rather
0.4 - 0.84 MU/kg/day (4 - 8.4 μg/kg/day) was used.
Patients treated with myeloablative therapy followed by bone marrow transplantation
Filgrastim is administered as an intravenous short-term infusion over 30 minutes or as a subcutaneous
or intravenous continuous infusion over 24 hours, in each case after dilution in 20 ml of glucose
50 mg/ml (5%) solution. For further instructions on dilution with glucose 50 mg/ml (5%) solution
prior to infusion see section 6.6.
Mobilisation of PBPC
Subcutaneous injection.
For the mobilisation of PBPC in patients undergoing myelosuppressive or myeloablative therapy
followed by autologous PBPC transplantation the recommended dose of filgrastim may also be
administered as a 24 hour subcutaneous continuous infusion. For infusions filgrastim should be diluted
in 20 ml of glucose 50 mg/ml (5%) solution. For further instructions on dilution with glucose
50 mg/ml (5%) solution prior to infusion see section 6.6.
SCN/HIV infection
Subcutaneous injection.
Hypersensitivity to the active substance or to any of the excipients.
Special warnings
Filgrastim should not be used to increase the dose of cytotoxic chemotherapy beyond established
posology regimens (see below).
21
Filgrastim should not be administered to patients with severe congenital neutropenia (Kostmann's
syndrome) with abnormal cytogenetics (see below).
Established cytotoxic chemotherapy
Malignant cell growth
Since investigations showed that G-CSF can promote growth of myeloid cells
in vitro
the following
warnings should be considered.
The safety and efficacy of filgrastim administration in patients with myelodysplastic syndrome, or
chronic myelogenous leukaemia have not been established. Therefore filgrastim is not indicated for
use in these conditions. Particular care should be taken to distinguish the diagnosis of blast
transformation of chronic myeloid leukaemia from acute myeloid leukaemia.
In view of limited safety and efficacy data in patients with secondary AML, filgrastim should be
administered with caution.
The safety and efficacy of filgrastim administration in
de novo
AML patients aged < 55 years with
good cytogenetics [t(8;21), t(15;17), and inv(16)] have not been established.
Leucocytosis
White blood cell counts of 100 x 10
9
/l or greater have been observed in less than 5% of patients
receiving filgrastim at doses above 0.3 MU/kg/day (3 μg/kg/day). No undesirable effects directly
attributable to this degree of leucocytosis have been reported. However, in view of the potential risks
associated with severe leucocytosis, a white blood cell count should be performed at regular intervals
during filgrastim therapy. If leukocyte counts exceed 50 x 10
9
/l after the expected nadir, filgrastim
should be discontinued immediately. However, during the period of administration of filgrastim for
PBPC mobilisation, it should be discontinued or its dose should be reduced if the leukocyte counts rise
to > 70 x 10
9
/l.
Risks associated with increased doses of chemotherapy
Special caution should be used when treating patients with high-dose chemotherapy because improved
tumour outcome has not been demonstrated and intensified doses of chemotherapeutic agents may lead
to increased toxicities including cardiac, pulmonary, neurologic, and dermatologic effects (please refer
to the Summary of Product Characteristics of the specific chemotherapy agents used).
Treatment with filgrastim alone does not preclude thrombocytopenia and anaemia due to
myelosuppressive chemotherapy. Because of the potential of receiving higher doses of chemotherapy
(e.g. full doses on the prescribed schedule) the patient may be at greater risk of thrombocytopenia and
anaemia. Regular monitoring of platelet count and haematocrit is recommended. Special care should
be taken when administering single or combination chemotherapeutic agents which are known to
cause severe thrombocytopenia.
The use of filgrastim-mobilised PBPCs has been shown to reduce the depth and duration of
thrombocytopenia following myelosuppressive or myeloablative chemotherapy.
Other special precautions
The effects of filgrastim in patients with substantially reduced myeloid progenitors have not been
studied. Filgrastim acts primarily on neutrophil precursors to exert its effect in elevating neutrophil
counts. Therefore, in patients with reduced precursors, neutrophil response may be diminished (such
as those treated with extensive radiotherapy or chemotherapy, or those with bone marrow infiltration
by tumour).
There have been reports of Graft versus Host Disease (GvHD) and fatalities in patients receiving
G-CSF after allogeneic bone marrow transplantation (see section 5.1).
22
Mobilisation of PBPC
Prior exposure to cytotoxic agents
Patients who have undergone very extensive prior myelosuppressive therapy, followed by
administration of filgrastim for mobilisation of PBPCs, may not show sufficient mobilisation of these
blood cells to achieve the recommended minimum yield (≥ 2.0 x 10
6
CD34+ cells/kg) or acceleration
of platelet recovery to the same degree.
Some cytotoxic agents exhibit particular toxicities to the haematopoietic progenitor pool and may
adversely affect progenitor mobilisation. Agents such as melphalan, carmustine (BCNU) and
carboplatin, when administered over prolonged periods prior to attempts at progenitor mobilisation
may reduce progenitor yield. However, the administration of melphalan, carboplatin or BCNU
together with filgrastim has been shown to be effective for progenitor mobilisation. When a PBPC
transplantation is envisaged it is advisable to plan the stem cell mobilisation procedure early in the
treatment course of the patient. Particular attention should be paid to the number of progenitors
mobilised in such patients before the administration of high-dose chemotherapy. If yields are
inadequate, as measured by the criteria above, alternative forms of treatment not requiring progenitor
support should be considered.
Assessment of progenitor cell yields
In assessing the number of progenitor cells harvested in patients treated with filgrastim, particular
attention should be paid to the method of quantitation. The results of flow cytometric analysis of
CD34+ cell numbers vary depending on the precise methodology used and therefore,
recommendations of numbers based on studies in other laboratories need to be interpreted with
caution.
Statistical analysis of the relationship between the number of CD34+ cells re-infused and the rate of
platelet recovery after high-dose chemotherapy indicates a complex but continuous relationship. The
recommendation of a minimum yield of ≥ 2.0 x 10
6
CD34+ cells/kg is based on published experience
resulting in adequate haematologic reconstitution. Yields in excess of this minimum yield appear to
correlate with more rapid recovery, those below with slower recovery.
Normal donors prior to allogeneic PBPC transplantation
Mobilisation of PBPC does not provide a direct clinical benefit to normal donors and should only be
considered for the purposes of allogeneic stem cell transplantation.
PBPC mobilisation should be considered only in donors who meet normal clinical and laboratory
eligibility criteria for stem cell donation with special attention to haematological values and infectious
disease.
The safety and efficacy of filgrastim have not been assessed in normal donors < 16 years or
> 60 years.
Transient thrombocytopenia (platelets < 100 x 10
9
/l) following filgrastim administration and
leukapheresis was observed in 35% of subjects studied. Among these, two cases of platelets
< 50 x 10
9
/l were reported and attributed to the leukapheresis procedure. If more than one
leukapheresis is required, particular attention should be paid to donors with platelets < 100 x 10
9
/l
prior to leukapheresis; in general apheresis should not be performed if platelets < 75 x 10
9
/l.
Leukapheresis should not be performed in donors who are anticoagulated or who have known defects
in haemostasis.
Filgrastim administration should be discontinued or its posology should be reduced if the leukocyte
counts rise to > 70 x 10
9
/l.
Donors who receive G-CSFs for PBPC mobilisation should be monitored until haematological indices
return to normal.
23
Transient cytogenetic modifications have been observed in normal donors following G-CSF use. The
significance of these changes is unknown.
Long-term safety follow-up of donors is ongoing. Nevertheless, a risk of promotion of a malignant
myeloid clone can not be excluded. It is recommended that the apheresis centre perform a systematic
record and tracking of the stem cell donors for at least 10 years to ensure monitoring of long-term
safety.
Common but generally asymptomatic cases of splenomegaly and very rare cases of splenic rupture
have been reported in healthy donors and patients following administration of G-CSFs. Some cases of
splenic rupture were fatal. Therefore, spleen size should be carefully monitored (e.g. clinical
examination, ultrasound). A diagnosis of splenic rupture should be considered in donors and/or
patients reporting left upper abdominal pain or shoulder tip pain.
In normal donors, pulmonary adverse events (haemoptysis, pulmonary haemorrhage, pulmonary
infiltrates, dyspnoea and hypoxia) have been reported very rarely in post marketing experience. In
case of suspected or confirmed pulmonary adverse events, discontinuation of treatment with filgrastim
should be considered and appropriate medical care given.
Recipients of allogeneic PBPCs mobilised with filgrastim
Current data indicate that immunological interactions between the allogeneic PBPC graft and the
recipient may be associated with an increased risk of acute and chronic GvHD when compared with
bone marrow transplantation.
SCN
Blood cell counts
Platelet counts should be monitored closely, especially during the first few weeks of filgrastim
therapy. Consideration should be given to intermittent cessation or decreasing the dose of filgrastim in
patients who develop thrombocytopenia, i.e. platelets consistently < 100,000/mm
3
.
Other blood cell changes occur, including anaemia and transient increases in myeloid progenitors,
which require close monitoring of cell counts.
Transformation to leukaemia or myelodysplastic syndrome
Special care should be taken in the diagnosis of SCNs to distinguish them from other haematopoietic
disorders such as aplastic anaemia, myelodysplasia, and myeloid leukaemia. Complete blood cell
counts with differential and platelet counts, and an evaluation of bone marrow morphology and
karyotype should be performed prior to treatment.
There was a low frequency (approximately 3%) of myelodysplastic syndromes (MDS) or leukaemia in
clinical study patients with SCN treated with filgrastim. This observation has only been made in
patients with congenital neutropenia. MDS and leukaemias are natural complications of the disease
and are of uncertain relation to filgrastim therapy. A subset of approximately 12% of patients who had
normal cytogenetic evaluations at baseline were subsequently found to have abnormalities, including
monosomy 7, on routine repeat evaluation. If patients with SCN develop abnormal cytogenetics, the
risks and benefits of continuing filgrastim should be carefully weighed; Filgrastim should be
discontinued if MDS or leukaemia occurs. It is currently unclear whether long-term treatment of
patients with SCN will predispose patients to cytogenetic abnormalities, MDS or leukaemic
transformation. It is recommended to perform morphologic and cytogenetic bone marrow
examinations in patients at regular intervals (approximately every 12 months).
Other special precautions
Causes of transient neutropenia, such as viral infections should be excluded.
Splenic enlargement is a direct effect of treatment with filgrastim. 31% of patients in studies were
documented as having palpable splenomegaly. Increases in volume, measured radiographically,
occurred early during Filgrastim therapy and tended to plateau. Dose reductions were noted to slow or
24
stop the progression of splenic enlargement, and in 3% of patients a splenectomy was required. Spleen
size should be evaluated regularly. Abdominal palpation should be sufficient to detect abnormal
increases in splenic volume.
Haematuria/proteinuria occurred in a small number of patients. Regular urine analyses should be
performed to monitor this event.
The safety and efficacy in neonates and patients with autoimmune neutropenia have not been
established.
HIV infection
Blood cell counts
ANC should be monitored closely, especially during the first few weeks of filgrastim therapy. Some
patients may respond very rapidly and with a considerable increase in neutrophil count to the initial
dose of filgrastim. It is recommended that the ANC is measured daily for the first 2 - 3 days of
filgrastim administration. Thereafter, it is recommended that the ANC is measured at least twice per
week for the first 2 weeks and subsequently once per week or once every other week during
maintenance therapy. During intermittent dosing with 30 MU/day (300 μg/day) of filgrastim, there can
be wide fluctuations in the patient's ANC over time. In order to determine a patient's trough or nadir
ANC, it is recommended that blood samples are taken for ANC measurement immediately prior to any
scheduled dosing with filgrastim.
Risk associated with increased doses of myelosuppressive medicinal products
Treatment with filgrastim alone does not preclude thrombocytopenia and anaemia due to
myelosuppressive treatments. As a result of the potential to receive higher doses or a greater number
of these medicinal products with filgrastim therapy, the patient may be at higher risk of developing
thrombocytopenia and anaemia. Regular monitoring of blood counts is recommended (see above).
Infections and malignancies causing myelosuppression
Neutropenia may be due to bone marrow-infiltrating opportunistic infections such as Mycobacterium
avium complex or malignancies such as lymphoma. In patients with known bone marrow-infiltrating
infections or malignancy, consider appropriate therapy for treatment of the underlying condition in
addition to administration of filgrastim for treatment of neutropenia. The effects of filgrastim on
neutropenia due to bone marrow-infiltrating infection or malignancy have not been well established.
Other special precautions
Rare pulmonary adverse reactions, in particular interstitial pneumonia, have been reported after
G-CSF administration (see section 4.8). Patients with a recent history of pulmonary infiltrates or
pneumonia may be at higher risk. The onset of pulmonary signs, such as cough, fever and dyspnoea in
association with radiological signs of pulmonary infiltrates and deterioration in pulmonary function
may be preliminary signs of Adult Respiratory Distress Syndrome (ARDS). Filgrastim should be
discontinued and appropriate treatment given in these cases.
Monitoring of bone density may be indicated in patients with underlying osteoporotic bone diseases
who undergo continuous therapy with filgrastim for more than 6 months.
Sickle cells crises, in some cases fatal, have been reported with the use of filgrastim in subjects with
sickle cell disease. Physicians should exercise caution when considering the use of filgrastim in
patients with sickle cell disease, and only after careful evaluation of the potential risks and benefits.
Increased haematopoietic activity of the bone marrow in response to growth factor therapy has been
associated with transient positive bone-imaging findings. This should be considered when interpreting
bone imaging results.
25
Excipients
Zarzio contains sorbitol. Patients with rare hereditary problems of fructose intolerance should not use
this medicinal product.
The safety and efficacy of filgrastim given on the same day as myelosuppressive cytotoxic
chemotherapy have not been definitively established. In view of the sensitivity of rapidly dividing
myeloid cells to myelosuppressive cytotoxic chemotherapy, the use of filgrastim is not recommended
in the period from 24 hours before to 24 hours after chemotherapy. Preliminary evidence from a small
number of patients treated concomitantly with filgrastim and 5-fluorouracil indicates that the severity
of neutropenia may be exacerbated.
Possible interactions with other haematopoietic growth factors and cytokines have not yet been
investigated in clinical studies.
Since lithium promotes the release of neutrophils, lithium is likely to potentiate the effect of filgrastim.
Although this interaction has not been formally investigated, there is no evidence that such an
interaction is harmful.
There are no adequate data from the use of filgrastim in pregnant women. There are reports in the
literature where the transplacental passage of filgrastim in pregnant women has been demonstrated.
There is no evidence from studies in rats and rabbits that filgrastim is teratogenic. An increased
incidence of embryo-loss has been observed in rabbits, but no malformation has been seen.
In pregnancy, the possible risk of filgrastim use to the foetus must be weighed against the expected
therapeutic benefit.
It is not known whether filgrastim is excreted in human milk, therefore it is not recommended for use
in breast-feeding women.
Filgrastim has no influence on the ability to drive and use machines.
The most commonly reported adverse reactions to filgrastim are mild to moderate musculoskeletal
pain occurring in more than 10% of patients. Musculoskeletal pain is usually controlled with standard
analgesics.
Adverse reactions listed below are classified according to frequency and system organ class.
Frequency groupings are defined according to the following convention: Very common (≥ 1/10),
common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very
rare (< 1/10,000), not known (cannot be estimated from the available data).
26
Table 1. Adverse reactions in clinical trials in cancer patients
Immune system disorders
Very rare:
Allergic-type reactions*, including anaphylaxis, skin rash, urticaria,
angioedema, dyspnoea and hypotension
Vascular disorders
Uncommon: Hypotension (transient)
Rare: Vascular disorders* including venoocclusive disease and fluid volume
disturbances
Respiratory, thoracic and mediastinal disorders
Very rare: Pulmonary oedema*, interstitial pneumonia*, pulmonary infiltrates*
Skin and subcutaneous tissue disorders
Very rare: Sweet's Syndrome*, cutaneous vasculitis
Musculoskeletal, connective tissue and bone disorders
Very common: Musculoskeletal pain (mild or moderate)
Common: Musculoskeletal pain (severe)
Very rare: Rheumatoid arthritis exacerbation
Renal and urinary disorders
Very rare:
Micturition disorders (predominantly dysuria)
Investigations
Very common: Blood alkaline phosphatase, blood lactate dehydrogenase (LDH),
gamma-glutamyltransferase (GGT) and blood uric acid increased
(reversible, dose-dependent, mild or moderate)
* see below
Table 2. Adverse reactions in clinical trials in normal donors undergoing PBPC mobilisation
Immune system disorders
Uncommon: Severe allergic reaction: anaphylaxis, angioedema, urticaria, rash
Blood and the lymphatic system disorders
Very common: Leucocytosis (WBC > 50 x 10
9
/l), thrombocytopenia (platelets
< 100 x 10
9
/l; transient)
Common: Splenomegaly (generally asymptomatic, also in patients)
Uncommon: Spleen disorder
Very rare: Splenic rupture (also in patients)
Nervous system disorders
Very common: Headache
Respiratory, thoracic and mediastinal disorders
Very rare
:
Haemoptysis*, pulmonary haemorrhage*, pulmonary infiltrates*,
dyspnoea*, hypoxia*
Musculoskeletal, connective tissue and bone disorders
Very common: Musculoskeletal pain (mild to moderate, transient)
Uncommon: Rheumatoid arthritis and arthritic symptoms exacerbation
Investigations
Common: Blood alkaline phosphatase and LDH increased (transient, minor)
Uncommon: Aspartate aminotransferase (AST) and blood uric acid increased
(transient, minor)
* see below
27














Table 3. Adverse reactions in clinical trials in SCN patients
Blood and the lymphatic system disorders
Very common: Anaemia, splenomegaly (may be progressive in a minority of cases)
Common: Thrombocytopenia
Uncommon: Spleen disorder
Nervous system disorders
Common: Headache
Respiratory, thoracic and mediastinal disorders
Very common: Epistaxis
Gastrointestinal disorders
Common: Diarrhoea
Hepato-biliary disorders
Common: Hepatomegaly
Skin and subcutaneous tissue disorders
Common: Cutaneous vasculitis (during long term use), alopecia, rash
Musculoskeletal, connective tissue and bone disorders
Very common: General musculoskeletal pain, bone pain
Common: Osteoporosis, arthralgia
Renal and urinary disorders
Uncommon: Haematuria, proteinuria
General disorders and administration site conditions
Common:
Investigations
Very common: Blood alkaline phosphatase, LDH and blood uric acid increased
(transient), blood glucose decreased (transient, moderate)
Table 4. Adverse reactions in clinical trials in HIV Patients
Blood and lymphatic system disorders
Common: Spleen disorder, splenomegaly*
Musculoskeletal, connective tissue and bone disorders
Very common: Musculoskeletal pain (mild to moderate)
* see below
In randomised, placebo-controlled clinical studies, filgrastim did not increase the incidence of
frequency in patients treated with filgrastim/chemotherapy and placebo/chemotherapy included nausea
and vomiting, alopecia, diarrhoea, fatigue, anorexia, mucositis, headache, cough, skin rash, chest pain,
generalised weakness, sore throat, constipation and unspecified pain.
Allergic reactions occurred on initial or subsequent treatment in patients receiving filgrastim. Overall,
reports were more common after intravenous administration. In some cases, symptoms have recurred
with rechallenge, suggesting a causal relationship. Filgrastim should be permanently discontinued in
patients who experience a serious allergic reaction.
Vascular disorders have been reported in patients undergoing high dose chemotherapy followed by
autologous bone marrow transplantation. The causal association with filgrastim has not been
established.
Pulmonary adverse reactions have in some cases been reported with an outcome of respiratory failure
or adult respiratory distress syndrome (ARDS), which may be fatal. In normal donors, pulmonary
adverse events (haemoptysis, pulmonary haemorrhage, pulmonary infiltrates, dyspnoea and hypoxia)
have been reported very rarely in post marketing experience (see section 4.4).
The occurrence of Sweet's syndrome (acute febrile neutrophilic dermatosis) has been reported
occasionally in cancer patients. However, since a significant percentage of these patients were
28
Injection site pain













suffering from leukaemia, a condition known to be associated with Sweet's syndrome, a causal
relationship with filgrastim has not been established.
Isolated cases of sickle cells crises have been reported in patients with sickle cell disease (see section
4.4). The frequency is not known.
In all cases of splenic enlargement in HIV patients this was mild or moderate on physical examination
and the clinical course was benign; no patients had a diagnosis of hypersplenism and no patients
underwent splenectomy. As splenic enlargement is a common finding in patients with HIV infection
and is present to varying degrees in most patients with AIDS, the relationship to filgrastim treatment is
unclear.
Immunogenicity
In four clinical studies none of the healthy volunteers or cancer patients developed anti-rhG-CSF
antibodies (neither binding nor neutralising) upon treatment with Zarzio.
The effects of filgrastim overdose have not been established.
5.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Colony stimulating factors, ATC Code: L03AA02
Human G-CSF is a glycoprotein which regulates the production and release of functional neutrophils
from the bone marrow. Zarzio containing r-metHuG-CSF (filgrastim) causes marked increases in
peripheral blood neutrophil counts within 24 hours, with minor increases in monocytes. In some SCN
patients filgrastim can also induce a minor increase in the number of circulating eosinophils and
basophils relative to baseline; some of these patients may present with eosinophilia or basophilia
already prior to treatment. Elevations of neutrophil counts are dose-dependent at recommended doses.
Neutrophils produced in response to filgrastim show normal or enhanced function as demonstrated by
tests of chemotactic and phagocytic function. Following termination of filgrastim therapy, circulating
neutrophil counts decrease by 50% within 1 - 2 days, and to normal levels within 1 - 7 days. As with
other haematopoietic growth factors, G-CSF has shown
in vitro
stimulating properties on human
endothelial cells, which have specific receptors for G-CSF. Accordingly, G-CSF has been shown to
induce endothelial cell functions related to angiogenesis. Additionally, G-CSF has been shown to
increase neutrophil migration across the vascular endothelium.
Use of filgrastim in patients undergoing cytotoxic chemotherapy
leads to significant reductions in the
incidence, severity and duration of neutropenia and febrile neutropenia. Treatment with filgrastim
significantly reduces the duration of febrile neutropenia, antibiotic use and hospitalisation after
induction chemotherapy for acute myelogenous leukaemia or myeloablative therapy followed by bone
marrow transplantation. The incidence of fever and documented infections were not reduced in either
setting. The duration of fever was not reduced in patients undergoing myeloablative therapy followed
by bone marrow transplantation.
Use of filgrastim, either alone, or after chemotherapy,
mobilises haematopoietic progenitor cells into
peripheral blood. These autologous PBPCs may be harvested and infused after high-dose cytotoxic
therapy, either in place of, or in addition to bone marrow transplantation. Infusion of PBPCs
accelerates haematopoietic recovery reducing the duration of risk for haemorrhagic complications and
the need for platelet transfusions.
One retrospective European study evaluating the use of G-CSF after allogeneic bone marrow
transplantation in patients with acute leukaemias suggested an increase in the risk of GvHD, treatment
29
related mortality (TRM) and mortality when G-CSF was administered. In a separate retrospective
international study in patients with acute and chronic myelogenous leukaemias, no effect on the risk of
GvHD, TRM and mortality was seen . A meta-analysis of allogeneic transplant studies, including the
results of nine prospective randomized trials, 8 retrospective studies and 1 case-controlled study, did
not detect an effect on the risks of acute GvHD, chronic GvHD or early treatment-related mortality.
Relative Risk (95% CI) of GvHD and TRM
Following treatment with G-CSF after bone marrow transplantation
Publication
Period of
Study
N
Acute Grade
II - IV GvHD
Chronic
GvHD
TRM
Meta-Analysis
(2003)
1986 - 2001
a
1198
1.08
(0.87, 1.33)
1.02
(0.82, 1.26)
0.70
(0.38, 1.31)
European
Retrospective
Study (2004)
1992 - 2002
b
1789
1.33
(1.08, 1.64)
1.29
(1.02, 1.61)
1.73
(1.30, 2.32)
1.26
(0.95, 1.67)
a
Analysis includes studies involving BM transplant during this period; some studies used GM-CSF
b
Analysis includes patients receiving BM transplant during this period
1995 - 2000
b
2110
1.11
(0.86, 1.42)
1.10
(0.86, 1.39)
Use of filgrastim for the mobilisation of PBPCs in normal donors prior to allogeneic PBPC
transplantation
In normal donors, a 1 MU/kg/day (10 μg/kg/day) dose administered subcutaneously for
4 - 5 consecutive days allows a collection of ≥ 4 x 10
6
CD34+ cells/kg recipient BW in the majority of
the donors after two leukaphereses.
Recipients of allogeneic PBPCs mobilised with filgrastim experienced significantly more rapid
haematological recovery, leading to a significant decrease in time to unsupported platelet recovery
when compared with allogeneic bone marrow transplantation.
Use of filgrastim in children or adults with SCN
(severe congenital, cyclic, and idiopathic neutropenia)
induces a sustained increase in ACNs in peripheral blood and a reduction of infection and related
events.
Use of filgrastim in patients with HIV infection
maintains normal neutrophil counts to allow scheduled
dosing of antiviral and/or other myelosuppressive treatments. There is no evidence that patients with
HIV infection treated with filgrastim show an increase in HIV replication.
5.2 Pharmacokinetic properties
Randomised, double-blind, single and multiple dose, crossover studies in 146 healthy volunteers
showed that the pharmacokinetic profile of Zarzio was comparable to that of the reference product
after subcutaneous and intravenous administration.
Absorption
A single subcutaneous dose of 0.5 MU/kg (5 µg/kg) resulted in maximum serum concentrations after a
t
max
of 4.5 ± 0.9 hours (mean ± SD).
Distribution
The volume of distribution in blood is approximately 150 ml/kg. Following subcutaneous
administration of recommended doses, serum concentrations were maintained above 10 ng/ml for
8 - 16 hours. There is a positive linear correlation between the dose and the serum concentration of
filgrastim, whether administered intravenously or subcutaneously.
30
International
Retrospective
Study (2006)












Elimination
The elimination of filgrastim is non-linear with respect to dose, the serum clearance decreases with
increasing dose. Filgrastim appears to be mainly eliminated by neutrophil mediated clearance, which
becomes saturated at higher doses. However, the serum clearance increases with repeated dosing while
the neutrophil count increases. The median serum elimination half-life (t
1/2
) of filgrastim after single
subcutaneous doses ranged from 2.7 hours (1.0 MU/kg, 10 µg/kg) to 5.7 hours (0.25 MU/kg,
2.5 µg/kg) and was prolonged after 7 days of dosing to 8.5 - 14 hours, respectively.
Continuous infusion with filgrastim over a period of up to 28 days, in patients recovering from
autologous bone-marrow transplantation, resulted in no evidence of drug accumulation and
comparable elimination half-lives.
5.3 Preclinical safety data
There are no preclinical data of relevance to the prescriber which are additional to that already
included in other sections of the Summary of Product Characteristics.
6.
6.1 List of excipients
Glutamic acid
Sorbitol (E420)
Polysorbate 80
Water for injections
6.2 Incompatibilities
Zarzio must not be diluted with sodium chloride solutions.
This medicinal product must not be mixed with other medicinal products except those mentioned in
section 6.6.
Diluted filgrastim may be adsorbed to glass and plastic materials, unless it is diluted in
glucose 50 mg/ml (5%) solution (see section 6.6).
6.3 Shelf life
30 months.
After dilution: Chemical and physical in-use stability of the diluted solution for infusion has been
demonstrated for 24 hours at 2°C to 8°C. From a microbiological point of view, the product should be
used immediately. If not used immediately, in-use storage times and conditions prior to use are the
responsibility of the user and would normally not be longer than 24 hours at 2°C to 8°C, unless
dilution has taken place in controlled and validated aseptic conditions.
6.4 Special precautions for storage
Store in a refrigerator (2°C - 8°C).
Keep the pre-filled syringe in the outer carton in order to protect from light.
Within its shelf-life and for the purpose of ambulatory use, the patient may remove the product from
the refrigerator and store it at room temperature (not above 25°C) for one single period of up to 72
hours. At the end of this period, the product should not be put back in the refrigerator and should be
disposed of.
31
For storage conditions of the diluted medicinal product, see section 6.3.
6.5 Nature and contents of container
Pre-filled syringe (type I glass) with injection needle (stainless steel), with or without a needle safety
guard, containing 0.5 ml solution.
Pack sizes of 1, 3, 5 or 10 pre-filled syringes.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
The solution should be visually inspected prior to use. Only clear solutions without particles should be
used. Accidental exposure to freezing temperatures does not adversely affect the stability of Zarzio.
Zarzio contains no preservative: In view of the possible risk of microbial contamination, Zarzio
syringes are for single use only.
Dilution prior to administration (optional)
If required, Zarzio may be diluted in glucose 50 mg/ml (5%) solution.
Dilution to a final concentration < 0.2 MU/ml (2 μg/ml) is not recommended at any time.
For patients treated with filgrastim diluted to concentrations < 1.5 MU/ml (15 μg/ml), human serum
albumin (HSA) should be added to a final concentration of 2 mg/ml.
Example: In a final volume of 20 ml, total doses of filgrastim less than 30 MU (300 μg) should be
given with 0.2 ml of human serum albumin 200 mg/ml (20%) solution Ph. Eur. added.
When diluted in glucose 50 mg/ml (5%) solution, filgrastim is compatible with glass and a variety of
plastics including polyvinylchloride, polyolefin (a copolymer of polypropylene and polyethylene) and
polypropylene.
Using the pre-filled syringe with a needle safety guard
The needle safety guard covers the needle after injection to prevent needle stick injury. This does not
affect normal operation of the syringe. Depress the plunger slowly and evenly until the entire dose has
been given and the plunger cannot be depressed any further. While maintaining pressure on the
plunger, remove the syringe from the patient. The needle safety guard will cover the needle when
releasing the plunger.
Using the pre-filled syringe without a needle safety guard
Administer the dose as per standard protocol.
Disposal
Any unused product or waste material should be disposed of in accordance with local requirements.
7.
Sandoz GmbH
Biochemiestrasse 10
A-6250 Kundl
Austria
32
8.
EU/1/08/495/005
EU/1/08/495/006
EU/1/08/495/007
EU/1/08/495/008
EU/1/08/495/013
EU/1/08/495/014
EU/1/08/495/015
EU/1/08/495/016
9.
06/02/2009
Detailed information on this medicinal product is available on the website of the European Medicines
Agency (EMA)
http://www.ema.europa.eu/
.
33
ANNEX II
A.
MANUFACTURER OF THE BIOLOGICAL ACTIVE SUBSTANCE AND
MANUFACTURING AUTHORISATION HOLDER RESPONSIBLE FOR
BATCH RELEASE
B.
CONDITIONS OF THE MARKETING AUTHORISATION
34
A. MANUFACTURER OF THE BIOLOGICAL ACTIVE SUBSTANCE(S) AND
MANUFACTURING AUTHORISATION HOLDERS RESPONSIBLE FOR BATCH
RELEASE
Name and address of the manufacturer(s) of the biological active substance
Sandoz GmbH
Biochemiestrasse 10
6250 Kundl
Austria
Name and address of the manufacturer(s) responsible for batch release
Sandoz GmbH
Biochemiestrasse 10
AT-6250 Kundl
Austria
B. CONDITIONS OF THE MARKETING AUTHORISATION
•
Medicinal product subject to restricted medical prescription (See Annex I: Summary of Product
Characteristics, 4.2).
•
CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND
EFFECTIVE USE OF THE MEDICINAL PRODUCT
Not applicable
•
OTHER CONDITIONS
Pharmacovigilance system
The MAH must ensure that the system of pharmacovigilance, as presented in Module 1.8.1. of the
Marketing Authorisation, is in place and functioning before and whilst the product is on the market.
Risk Management Plan
The MAH commits to performing the studies and additional pharmacovigilance activities detailed in
the Pharmacovigilance Plan, as agreed in version 4.0, dated 20 October 2008, of the Risk Management
Plan (RMP) presented in Module 1.8.2. of the Marketing Authorisation Application and any
subsequent updates of the RMP agreed by the CHMP.
As per the CHMP Guideline on Risk Management Systems for medicinal products for human use, the
updated RMP should be submitted at the same time as the next Periodic Safety Update Report
(PSUR).
In addition, an updated RMP should be submitted
•
When new information is received that may impact on the current Safety Specification,
Pharmacovigilance Plan or risk minimisation activities
35
•
Within 60 days of an important (pharmacovigilance or risk minimisation) milestone being
reached
•
At the request of the EMA
36
ANNEX III
LABELLING AND PACKAGE LEAFLET
37
A. LABELLING
38
PARTICULARS TO APPEAR ON THE OUTER PACKAGING
OUTER CARTON – PRE-FILLED SYRINGE
1.
Zarzio 30 MU/0.5 ml solution for injection or infusion in pre-filled syringe
Filgrastim
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each pre-filled syringe contains 30 million units (equivalent to 300 micrograms) filgrastim in 0.5 ml
(60 MU/ml).
3.
LIST OF EXCIPIENTS
Excipients: glutamic acid, polysorbate 80, water for injections and sorbitol (E420) - see the package
leaflet for further information.
4.
Solution for injection or infusion in pre-filled syringe
1 pre-filled syringe without needle safety guard
3 pre-filled syringes without needle safety guard
5 pre-filled syringes without needle safety guard
10 pre-filled syringes without needle safety guard
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Single use only. Read the package leaflet before use.
Subcutaneous or intravenous use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
After dilution use within 24 hours.
39





















9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator (2°C - 8°C).
Keep the pre-filled syringe in the outer carton in order to protect from light.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Sandoz GmbH
Biochemiestrasse 10
A-6250 Kundl
Austria
EU/1/08/495/009
EU/1/08/495/010
EU/1/08/495/011
EU/1/08/495/012
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Zarzio 30 MU/0.5 ml
40





















PARTICULARS TO APPEAR ON THE OUTER PACKAGING
OUTER CARTON – PRE-FILLED SYRINGE
1.
Zarzio 48 MU/0.5 ml solution for injection or infusion in pre-filled syringe
Filgrastim
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each pre-filled syringe contains 48 million units (equivalent to 480 micrograms) filgrastim in 0.5 ml
(96 MU/ml).
3.
LIST OF EXCIPIENTS
Excipients: glutamic acid, polysorbate 80, water for injections and sorbitol (E420) - see the package
leaflet for further information.
4.
Solution for injection or infusion in pre-filled syringe
1 pre-filled syringe without needle safety guard
3 pre-filled syringes without needle safety guard
5 pre-filled syringes without needle safety guard
10 pre-filled syringes without needle safety guard
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Single use only. Read the package leaflet before use.
Subcutaneous or intravenous use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
After dilution use within 24 hours.
41





















9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator (2°C - 8°C).
Keep the pre-filled syringe in the outer carton in order to protect from light.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Sandoz GmbH
Biochemiestrasse 10
A-6250 Kundl
Austria
EU/1/08/495/013
EU/1/08/495/014
EU/1/08/495/015
EU/1/08/495/016
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Zarzio 48 MU/0.5 ml
42





















PARTICULARS TO APPEAR ON THE OUTER PACKAGING
OUTER CARTON – PRE-FILLED SYRINGE WITH A NEEDLE SAFETY GUARD
1.
Zarzio 30 MU/0.5 ml solution for injection or infusion in pre-filled syringe
Filgrastim
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each pre-filled syringe contains 30 million units (equivalent to 300 micrograms) filgrastim in 0.5 ml
(60 MU/ml).
3.
LIST OF EXCIPIENTS
Excipients: glutamic acid, polysorbate 80, water for injections and sorbitol (E420) - see the package
leaflet for further information.
4.
Solution for injection or infusion in pre-filled syringe
1 pre-filled syringe with a needle safety guard
3 pre-filled syringes with a needle safety guard
5 pre-filled syringes with a needle safety guard
10 pre-filled syringes with a needle safety guard
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Single use only. Read the package leaflet before use.
Subcutaneous or intravenous use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
After dilution use within 24 hours.
43





















9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator (2°C - 8°C).
Keep the pre-filled syringe in the outer carton in order to protect from light.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Sandoz GmbH
Biochemiestrasse 10
A-6250 Kundl
Austria
EU/1/08/495/001
EU/1/08/495/002
EU/1/08/495/003
EU/1/08/495/004
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Zarzio 30 MU/0.5 ml
44





















PARTICULARS TO APPEAR ON THE OUTER PACKAGING
OUTER CARTON – PRE-FILLED SYRINGE WITH A NEEDLE SAFETY GUARD
1.
Zarzio 48 MU/0.5 ml solution for injection or infusion in pre-filled syringe
Filgrastim
2.
STATEMENT OF ACTIVE SUBSTANCE(S)
Each pre-filled syringe contains 48 million units (equivalent to 480 micrograms) filgrastim in 0.5 ml
(96 MU/ml).
3.
LIST OF EXCIPIENTS
Excipients: glutamic acid, polysorbate 80, water for injections and sorbitol (E420) - see the package
leaflet for further information.
4.
Solution for injection or infusion in pre-filled syringe
1 pre-filled syringe with a needle safety guard
3 pre-filled syringes with a needle safety guard
5 pre-filled syringes with a needle safety guard
10 pre-filled syringes with a needle safety guard
5.
METHOD AND ROUTE(S) OF ADMINISTRATION
Single use only. Read the package leaflet before use.
Subcutaneous or intravenous use.
6.
SPECIAL WARNING THAT THE MEDICINAL PRODUCT MUST BE STORED OUT
OF THE REACH AND SIGHT OF CHILDREN
Keep out of the reach and sight of children.
7.
OTHER SPECIAL WARNING(S), IF NECESSARY
8.
EXPIRY DATE
EXP
After dilution use within 24 hours.
45





















9.
SPECIAL STORAGE CONDITIONS
Store in a refrigerator (2°C - 8°C).
Keep the pre-filled syringe in the outer carton in order to protect from light.
10. SPECIAL PRECAUTIONS FOR DISPOSAL OF UNUSED MEDICINAL PRODUCTS
OR WASTE MATERIALS DERIVED FROM SUCH MEDICINAL PRODUCTS, IF
APPROPRIATE
Sandoz GmbH
Biochemiestrasse 10
A-6250 Kundl
Austria
EU/1/08/495/005
EU/1/08/495/006
EU/1/08/495/007
EU/1/08/495/008
13. BATCH NUMBER
Lot
14. GENERAL CLASSIFICATION FOR SUPPLY
Medicinal product subject to medical prescription.
15. INSTRUCTIONS ON USE
16. INFORMATION IN BRAILLE
Zarzio 48 MU/0.5 ml
46





















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
PRE-FILLED SYRINGE / PRE-FILLED SYRINGE WITH A NEEDLE SAFETY GUARD
1.
Zarzio 30 MU/0.5 ml injection or infusion
Filgrastim
SC/IV
2.
METHOD OF ADMINISTRATION
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
0.5 ml
6.
OTHER
47
















MINIMUM PARTICULARS TO APPEAR ON SMALL IMMEDIATE PACKAGING UNITS
PRE-FILLED SYRINGE / PRE-FILLED SYRINGE WITH A NEEDLE SAFETY GUARD
1.
Zarzio 48 MU/0.5 ml injection or infusion
Filgrastim
SC/IV
2.
METHOD OF ADMINISTRATION
3.
EXPIRY DATE
EXP
4.
BATCH NUMBER
Lot
5.
CONTENTS BY WEIGHT, BY VOLUME OR BY UNIT
0.5 ml
6.
OTHER
48
















B. PACKAGE LEAFLET
49
PACKAGE LEAFLET: INFORMATION FOR THE USER
Zarzio 30 MU/0.5 ml solution for injection or infusion in pre-filled syringe
Zarzio 48 MU/0.5 ml solution for injection or infusion in pre-filled syringe
Filgrastim
Read all of this leaflet carefully before you start using this medicine.
-
Keep this leaflet. You may need to read it again.
-
If you have any further questions, ask your doctor or pharmacist.
-
This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even
if their symptoms are the same as yours.
-
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet,
please tell your doctor or pharmacist.
In this leaflet:
1.
What Zarzio is and what it is used for
2.
Before you use Zarzio
3.
How to use Zarzio
4.
Possible side effects
5.
How to store Zarzio
6.
Further information
1.
WHAT ZARZIO IS AND WHAT IT IS USED FOR
Zarzio contains the active substance filgrastim. It belongs to a group of proteins called cytokines, and
is very similar to the granulocyte-colony stimulating factor (G-CSF) produced by the human body.
Filgrastim stimulates the bone marrow to produce more white blood cells that help fight infection. If
the number of white blood cells is too low (neutropenia), the risk of infection increases.
Anticancer chemotherapy
Zarzio is used to reduce the duration of neutropenia and the occurrence of febrile neutropenia (with
fever) which can be caused by the use of cytotoxic anticancer chemotherapy in children and adults. It
is not used in patients with chronic myeloid leukaemia (CML) and myelodysplastic syndromes
(MDS).
Bone marrow transplantation
Zarzio is used to reduce the duration of neutropenia after high-dose chemotherapy and total body
irradiation (radiotherapy) followed by bone marrow transplantation, in children and adults at increased
risk of prolonged severe neutropenia.
Peripheral blood stem cell mobilisation
Zarzio is used to stimulate the bone marrow to release (mobilisation) peripheral blood progenitor cells
(PBPC, a type of stem cells) into the blood, so that they eventually grow and develop into all types of
blood cells: white blood cells, red blood cells and platelets.
If you are a patient with cancer these PBPCs will be removed from your blood and returned after your
chemotherapy and/or radiotherapy. As chemotherapy and/or radiotherapy can depress the activity of
your bone marrow, this will help to speed up its recovery.
If you donate stem cells for another person, the PBPCs will be removed from your blood and given to
the recipient after he/she has received their chemotherapy and/or radiotherapy.
50
Severe chronic neutropenia
Zarzio is used in children and adults to increase the number of white blood cells and reduce the
occurrence and duration of infections related to specific forms of severe chronic neutropenia:
congenital (inborn), cyclic (recurring), or idiopathic (without known cause).
HIV infection neutropenia
Zarzio is used to treat persistent neutropenia in patients with advanced HIV infection, in order to
reduce the risk of bacterial infections, when other treatments are not appropriate.
2.
BEFORE YOU USE ZARZIO
Do not use Zarzio
-
if you are allergic (hypersensitive) to filgrastim or to any of the other ingredients of Zarzio
(listed in section 6. ‘What Zarzio contains’).
Take special care with Zarzio
Please tell your doctor:
-
if you have sickle cell anaemia (disorder of the red blood cells leading to a sickle-shaped form
of the red blood cells).
-
if you have specific blood disorders, such as myelodysplastic syndromes (MDS) or chronic
myeloid leukaemia (CML).
-
if you act as a donor for another person and are treated with anticoagulants (blood thinning
medicines) or you have bleeding problems.
Using other medicines
Please tell your doctor or pharmacist if you are taking or have recently taken any other medicines,
including medicines obtained without a prescription.
Pregnancy and breast-feeding
It is important to tell your doctor if you are pregnant, think you may be pregnant or plan to get
pregnant, as the doctor may decide that you should not use this medicine. Filgrastim could affect your
ability to become or stay pregnant.
Your doctor may decide that you should not use filgrastim if you are breast-feeding, as it is not known
whether it passes into milk.
Ask your doctor or pharmacist for advice before taking any medicine.
Driving and using machines
Zarzio has no influence on the ability to drive and use machines.
Important information about some of the ingredients of Zarzio
This medicine contains sorbitol (a type of sugar). If you have been told by your doctor that you have
an intolerance to some sugars, contact your doctor before taking this medicine.
3.
HOW TO USE ZARZIO
The amount of Zarzio you need depends on your bodyweight and the condition you are treated for.
Anticancer chemotherapy
The usual dose is 0.5 million units (MU) per kg bodyweight each day. For example, if you weigh
60 kg, your daily dose will be 30 MU. The first dose should not be administered less than 24 hours
51
-
if you have osteoporosis.
following cytotoxic chemotherapy. Your treatment can last up to 14 days. In some disease types,
however, longer treatment of up to about 1 month may be required.
Bone marrow transplantation
The usual starting dose is 1 MU per kg bodyweight each day. For example, if you weigh 60 kg, your
daily dose will be 60 MU. You will normally receive your first dose at least 24 hours after your
chemotherapy, but within 24 hours of receiving your bone marrow transfusion. Your doctor will then
test your blood to tell how well your treatment is working and how long it should last.
Peripheral blood stem cell mobilisation
-
If you are donating stem cells for yourself, the usual dose is 1 MU per kg bodyweight each day.
Zarzio will be given for 5 - 7 consecutive days. Treatment with Zarzio should be maintained
until the last stem cell collection.
-
If you are donating stem cells for yourself after chemotherapy, the usual dose is
0.5 MU per kg bodyweight each day. Zarzio will be given until the expected lowest level of
white blood cells is passed and has recovered to the normal range.
-
If you are acting as a stem cell donor for another person, the usual dose is
1 MU per kg bodyweight each day. Zarzio treatment will last for 4 - 5 consecutive days.
Your doctor will perform regular blood tests to determine the best time to collect the stem cells.
Severe chronic neutropenia
The usual starting dose is between 0.5 and 1.2 MU per kg bodyweight each day in a single or divided
dose. Your doctor will then test your blood to see how well your treatment is working and to find the
dose that is best for you. Long-term treatment with Zarzio is required for neutropenia.
HIV infection neutropenia
The usual starting dose is between 0.1 and 0.4 MU per kg bodyweight each day. Your doctor will test
your blood at regular intervals to see how well the treatment is working. Once the number of white
cells in your blood have returned to normal it may be possible to reduce the dose frequency to less
than once per day. Your doctor will continue to test your blood regularly and will recommend the best
dose for you. Long-term treatment with Zarzio may be required to maintain a normal number of white
cells in your blood.
Children and adolescents
The dose recommendations are the same as those for adults receiving chemotherapy.
How Zarzio is used
Always use Zarzio exactly as your doctor has told you. You should check with your doctor, nurse or
pharmacist if you are not sure. This medicine is given by injection into the tissue just under the skin
(subcutaneous injection), or through an intravenous infusion (“a drip”).
Your doctor may decide that it would be more convenient for you to inject Zarzio yourself. Your
doctor or nurse will show you how to inject yourself.
Do not try to inject yourself if you have not
been trained.
Instructions on how to inject yourself with Zarzio can be found at the end of this leaflet.
If you use more Zarzio than you should
If you have used more Zarzio than you should, contact your nurse, doctor or pharmacist immediately.
52
If you forget to use Zarzio
If you have forgotten a dose of Zarzio, you should contact your doctor to discuss when you should
inject the next dose.
If you stop using Zarzio
Your doctor will tell you when you can stop using Zarzio. It is quite normal to have several courses of
treatment with Zarzio.
4.
POSSIBLE SIDE EFFECTS
Like all medicines, Zarzio can cause side effects, although not everybody gets them.
Tell your doctor immediately
•
if you experience a cough, fever and difficulty breathing since you may develop severe pulmonary
side effects like pneumonia and respiratory distress;
•
if you get pain in the upper left side of your stomach or pain at the tip of your shoulder since this
may relate to a problem with your spleen;
•
if you have sudden difficulty breathing or dizziness, swelling of your face or throat, skin weals or
rash. As this may be a serious allergic reaction, stop your injection and get medical help
immediately.
Side effects may occur with certain frequencies, which are defined as follows:
•
very common: affects more than 1 user in 10
•
common: affects 1 to 10 users in 100
•
uncommon: affects 1 to 10 users in 1,000
•
rare: affects 1 to 10 users in 10,000
•
very rare: affects less than 1 user in 10,000
•
not known: frequency cannot be estimated from the available data.
Very common side effects
•
bone, joint and muscle pain
•
elevations in blood levels of uric acid and certain enzymes
•
decreases in blood glucose
•
leukocytosis (an abnormally high number of white blood cells in the blood)
•
low level of blood platelets (which increases risk of bleeding or bruising)
•
low level of red blood cells (which can make the skin pale and cause weakness and
breathlessness)
•
headache
•
nose bleed
•
enlarged spleen
Common side effects
•
diarrhoea
•
injection site pain
•
hair loss
•
rash
•
enlarged liver
•
inflammation of blood vessels of the skin
•
loss of calcium from the bones (osteoporosis)
•
spleen disorder
Uncommon side effects
•
decrease in blood pressure
•
allergic-type reactions, including anaphylaxis (dizziness, severe drop in blood pressure, difficulty
in breathing), angioedema (painful swelling of the face or throat), difficulty in breathing, low
53
blood pressure, skin rash, nettle rash; these reactions can occur at the start of the treatment or
throughout the treatment
•
worsening of rheumatoid arthritis or arthritic symptoms
•
blood or protein in the urine
Rare side effects
•
vascular disorders, including venoocclusive disease (a disease which affects the liver) and water
retention, which can cause swelling in the limbs
Very rare side effects
•
difficulties in passing urine
•
Sweet's syndrome (plum-coloured, raised, painful lesions on the limbs, sometimes the face and
neck, with fever)
•
water retention in the lungs, bleeding in the lungs, coughing up of blood, lack of oxygen,
inflammation of the lungs (which can cause breathlessness, cough, raised temperature); in some
cases, this could lead to respiratory failure or adult respiratory distress syndrome (ARDS, a
condition causing increased shortness of breath, in very ill patients), which may be fatal
•
sickle cell crises (acute collapse of red blood cells) in patients with sickle cell disease
•
rupture of the spleen
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet, please
tell your doctor or pharmacist.
5.
HOW TO STORE ZARZIO
Keep out of the reach and sight of children.
Do not use Zarzio after the expiry date which is stated on the carton and on the syringe label after
EXP. The expiry date refers to the last day of that month.
Store in a refrigerator (2°C - 8°C).
The syringe can be removed from the refrigerator and left at room temperature for a single period of
maximum 72 hours (but not above 25°C). At the end of this period, the product should not be put back
in the refrigerator and should be disposed of.
Keep the pre-filled syringe in the outer carton in order to protect from light.
Medicines should not be disposed of via wastewater or household waste. Ask your pharmacist how to
dispose of medicines no longer required. These measures will help to protect the environment.
6.
FURTHER INFORMATION
What Zarzio contains
-
The active substance is filgrastim.
Zarzio 30 MU/0.5 ml solution for injection or infusion in pre-filled syringe: Each pre-filled
syringe contains 30 MU filgrastim in 0.5 ml, corresponding to 60 MU/ml.
Zarzio 48 MU/0.5 ml solution for injection or infusion in pre-filled syringe: Each pre-filled
syringe contains 48 MU filgrastim in 0.5 ml, corresponding to 96 MU/ml
-
The other ingredients are glutamic acid, sorbitol (E420), polysorbate 80 and water for
injections.
54
What Zarzio looks like and contents of the pack
Zarzio is a clear, colourless to slightly yellowish solution for injection or infusion in a pre-filled
syringe.
Zarzio is available in packs containing 1, 3, 5 or 10 pre-filled syringes with injection needle and with
or without a needle safety guard.
Not all pack sizes may be marketed.
Marketing Authorisation Holder and Manufacturer
Sandoz GmbH
Biochemiestrasse 10
A-6250 Kundl
Austria
For any information about this medicinal product, please contact the local representative of the
Marketing Authorisation Holder:
België/Belgique/Belgien
Sandoz nv-sa
Telecom Gardens
Medialaan 40
B-1800 Vilvoorde
Tél/Tel: +32 27229797
Luxembourg/Luxemburg
Sandoz GmbH
Biochemiestr. 10
A-6250 Kundl
Tel: +43 5338 2000
България
Представителство Сандоз д.д.
Младост 4, Бизнес Парк София, Сграда 8Б,
ет.6
София 1715
Тел.: +359 2 970 47 47
Magyarország
Sandoz Hungária Kft.
Bartók Béla út 43.-47.
H-1114 Budapest
Tel.: +36 1 430 2890
info.hungary@sandoz.com
Česká republika
Sandoz s.r.o.
U Nákladového nádraží 10
CZ 130 00 Praha 3
Tel: +420 221 421 611
office.cz@sandoz.com
Malta
Sandoz GmbH
Biochemiestr. 10
A-6250 Kundl
Tel: +43 5338 2000
Danmark
Sandoz A/S
Edvard Thomsens Vej 14
DK-2300 K
øbenhavn S
Tlf: +45 6395 1000
Nederland
Sandoz B.V.
Veluwezoom 22
NL-1327 AH Almere
Tel: +31 36 52 41 600
55
Deutschland
NeoCorp AG
Am Weidenbach 6
D-82362 Weilheim
Tel: +49 881 909 5960
info@neocorp.de
oder
Sandoz Pharmaceuticals GmbH
Raiffeisenstraße 11
D-83607 Holzkirchen
Tel: +49 8024 902 4000
Norge
Sandoz A/S
Edvard Thomsens Vej 14
DK-2300 K
øbenhavn S
Tlf: +45 6395 1000
Eesti
Sandoz d.d. Eesti filiaal
Pärnu mnt 105
EE – 11312 Tallinn
Tel: +372 6652 400
Österreich
Sandoz GmbH
Biochemiestr. 10
A-6250 Kundl
Tel: +43 5338 2000
Ελλάδα
Novartis (Hellas) A.E.B.E.
12ο χλμ Εθνικής Οδού Νο.1
144 51 Μεταμόρφωση
Τηλ: +30 210 281 17 12
ή
DEMO Ανώνυμος Βιομηχανική και Εμπορική
Εταιρεία
21o χλμ. Εθν. Οδού Αθηνών-Λαμίας,
145 68, Αθήνα, Ελλάδα
Тел.: +210 8161802
Polska
Sandoz Polska Sp. z o.o.
ul. Domaniewska 50 C
PL – 02 672 Warszawa
Tel.: +48 22 549 15 00
España
Sandoz Farmacéutica, SA
Avda. Osa Mayor, 4
E-28023 Aravaca (Madrid)
Tel. +34 91 548 84 04
sandoz.responde@sandoz.com
Portugal
Sandoz Farmacêutica Lda.
Alameda da Beloura, Edifício 1
2° andar – Esc. 15
P-2710−693 Sintra
Tel: +351 21 924 19 11
sandoz.pt@sandoz.com
France
Sandoz SAS
49, avenue Georges Pompidou
F-92593 Levallois-Perret Cedex
Tél: +33 1 49 64 48 00
România
S.C. Sandoz SRL
Str. Livezeni, Nr 7A
Târgu Mureş, 540472 - RO
Tel: +40 265 208 120
Ireland
Rowex Ltd
Bantry
Co. Cork - IRL
Tel: +353 27 50077
reg@rowa-pharma.ie
Slovenija
Lek farmacevtska družba d.d.
Verovškova 57
SI-1526 Ljubljana
Tel: +386 1 580 21 11
56
Ísland
Sandoz A/S
Edvard Thomsens Vej 14
DK-2300 K
øbenhavn S
Tlf: +45 6395 1000
Slovenská republika
Sandoz d.d. - organizačná zložka
Galvaniho 15/C
SK-821 04 Bratislava
Tel: +421 2 48 200 600
Italia
Sandoz S.p.A.
Largo Umberto Boccioni, 1
I-21040 Origgio / VA
Tel: +39 02 96541
Suomi/Finland
Sandoz A/S
Edvard Thomsens Vej 14
DK-2300 K
ööpenhamina S
Puh/Tel: +45 6395 1000
Κύπρος
Διανοµέας Κύттου
Π.θ.Χαtζηγεωργίου ΣΙΑ ΛΤΔ
Гιλtίζ 31- 3042 Λεµεσός
Τηλέfωνο: +357 25372425
Sverige
Sandoz A/S
Edvard Thomsens Vej 14
DK-2300 K
örpenhamn S
Tel: +45 6395 1000
Latvija
Sandoz d.d. Pārstāvniecība Latvijā
K. Valdemāra iela 33 - 30
Rīga, LV-1010
Tel: +371 67 892 006
United Kingdom
Sandoz Limited
Frimley Business Park
Frimley, Camberley
Surrey GU16 7SR – UK
Tel: +44 1276 69 8020
uk.drugsafety@sandoz.com
Lietuva
Sandoz Pharmaceuticals d.d filialas
Šeimyniškių g. 3A
LT – 09312 Vilnius
Tel: +370 5 2636 038
This leaflet was last approved in
Detailed information on this medicine is available on the European Medicines Agency (EMA) web
site: http://www.ema.europa.eu/
-------------------------------------------------------------------------------------------------------------------------
Instructions on how to inject yourself
This section contains information on how to give yourself an injection of Zarzio.
It is important that
you do not try to give yourself the injection unless you have received special training from your
doctor or nurse.
Zarzio is provided with or without a needle safety guard and you will be shown how
to use this by your doctor or nurse. If you are not sure about giving the injection or you have any
questions, please ask your doctor or nurse for help.
How do I inject myself?
You will need to give yourself an injection into the tissue under the skin, known as a subcutaneous
injection. Your doctor or nurse will tell you how frequently it should be injected.
What do I need?
To give yourself a subcutaneous injection you will need:
1.
A new pre-filled syringe of Zarzio with or without a needle safety guard
57
2.
Alcohol wipes or similar
3.
A puncture-proof container for disposing of used syringes safely if you use the Zarzio pre-filled
syringe without a needle safety guard.
What should I do before I give myself a subcutaneous injection of Zarzio?
1.
Take your Zarzio pack out of the refrigerator. Accidental exposure to freezing temperatures
does not adversely affect the stability of Zarzio.
2.
Do not shake the packing containing the pre-filled syringe.
3.
Check that it is the correct strength you need for the dose that your doctor has prescribed.
4.
Check the expiry date which is stated on the carton and syringe label (EXP). Do not use it if the
date has passed the last day of the month shown.
5.
Check the appearance of Zarzio. It must be clear. If it is cloudy or there are particles in it, you
must not use it.
6.
For a more comfortable injection, let the pre-filled syringe stand for 30 minutes to reach room
temperature or hold the pre-filled syringe gently in your hand for a few minutes.
Do not warm
Zarzio in any other way
(for example, do not warm it in a microwave or in hot water).
7.
Do not
remove the plastic cover from the needle until you are ready to inject.
8.
Wash your hands thoroughly
.
9.
Find a comfortable, well-lit place and put the syringe, the alcohol wipes and if necessary the
puncture-proof container where you can reach them.
How do I prepare the injection?
Before you inject Zarzio you must do the following:
1.
Gently remove the plastic cover from the needle without twisting it. Do not touch the needle or
push the plunger.
2.
Hold the syringe with the needle pointing upwards to check if there are air bubbles in the
syringe. If there are air bubbles in it expel all air from the syringe by cautiously pushing the
plunger upwards (avoid expelling liquid from the syringe).
3.
The syringe has a scale on the syringe barrel. Push the plunger up to the number (ml) that
matches the dose of Zarzio that your doctor prescribed.
4.
Check again to make sure the correct dose of Zarzio is in the syringe.
5.
You can now use the pre-filled syringe.
Where should I give my injection?
The most suitable places to inject yourself are:
-
The top of your thighs; and
-
The abdomen,
except for the area around the navel.
Change the place that you inject each time so you don’t become sore in one area. If
someone is injecting for you, they can also use the back of your arms.
How do I give my injection?
1.
Disinfect your skin by using an alcohol wipe and pinch the skin between your thumb and
forefinger, without squeezing it.
2.
Insert the needle under the skin at an angle of approximately 45° as shown by your nurse or
doctor.
3.
Pull slightly on the plunger to check that a blood vessel has not been punctured. If you see
blood in the syringe, remove the needle and put it in another place.
Pre-filled syringe without needle safety guard
58
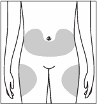
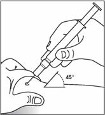
4.
Always keeping your skin pinched, depress the plunger slowly and evenly.
5.
After injecting the liquid, remove the needle and let go of your skin.
6.
Put the used syringe in the disposal container. Use each syringe only for one injection.
Pre-filled syringe with needle safety guard
4.
Always keeping your skin pinched, depress the plunger slowly and evenly
until the entire dose has been given and the plunger cannot be depressed any
further. Do not release the pressure on the plunger!
5.
After injecting the liquid, remove the needle while maintaining pressure on
the plunger and then let go of your skin.
6.
Let go of the plunger. The needle safety guard will rapidly move to cover the
needle.
7.
Use each syringe only for one injection.
Remember
If you have any problems, please do not be afraid to ask your doctor or nurse for help and advice.
Disposal of used syringes
The used syringes should be disposed of in accordance with local requirements.
Pre-filled syringe without needle safety guard
-
Do not put the cover back on used syringes.
-
Put used syringes into the puncture-proof disposal container and keep it out of the reach and
sight of children.
-
Dispose of the full container as instructed by your doctor, nurse or pharmacist.
-
NEVER
put used syringes into your normal household waste bin.
Pre-filled syringe with needle safety guard
-
The needle safety guard prevents needle stick injuries after use, so no special disposal
precautions are required. Dispose of the syringe as instructed by your doctor, nurse or
pharmacist.
-------------------------------------------------------------------------------------------------------------------------
The following information is intended for medical or healthcare professionals only:
The solution should be visually inspected prior to use. Only clear solutions without particles should be
used. Accidental exposure to freezing temperatures does not adversely affect the stability of Zarzio.
Zarzio contains no preservative: In view of the possible risk of microbial contamination, Zarzio
syringes are for single use only.
Dilution prior to administration (optional)
If required, Zarzio may be diluted in glucose 50 mg/ml (5%) solution. Zarzio must not be diluted with
sodium chloride solutions.
Dilution to a final concentration < 0.2 MU/ml (2 μg/ml) is not recommended at any time.
For patients treated with filgrastim diluted to concentrations < 1.5 MU/ml (15 μg/ml), human serum
albumin (HSA) should be added to a final concentration of 2 mg/ml.
Example: In a final volume of 20 ml, total doses of filgrastim less than 30 MU (300 μg) should be
given with 0.2 ml of human serum albumin 200 mg/ml (20%) solution Ph. Eur. added.
59

When diluted in glucose 50 mg/ml (5%) solution, filgrastim is compatible with glass and a variety of
plastics including polyvinylchloride, polyolefin (a copolymer of polypropylene and polyethylene) and
polypropylene.
After dilution: Chemical and physical in-use stability of the diluted solution for infusion has been
demonstrated for 24 hours at 2°C to 8°C. From a microbiological point of view, the product should be
used immediately. If not used immediately, in-use storage times and conditions prior to use are the
responsibility of the user and would normally not be longer than 24 hours at 2°C to 8°C, unless
dilution has taken place in controlled and validated aseptic conditions.
Using the pre-filled syringe with a needle safety guard
The needle safety guard covers the needle after injection to prevent needle stick injury. This does not
affect normal operation of the syringe. Depress the plunger slowly and evenly until the entire dose has
been given and the plunger cannot be depressed any further. While maintaining pressure on the
plunger, remove the syringe from the patient. The needle safety guard will cover the needle when
releasing the plunger.
Using the pre-filled syringe without a needle safety guard
Administer the dose as per standard protocol.
Disposal
Any unused product or waste material should be disposed of in accordance with local requirements.
60
Source: European Medicines Agency
 - Please bookmark this page (add it to your favorites).
- Please bookmark this page (add it to your favorites).
- If you wish to link to this page, you can do so by referring to the URL address below this line.
https://theodora.com/drugs/eu/zarzio.html
Copyright © 1995-2021 ITA all rights reserved.