| DRUGS INDEX | MANUFACTURERS INDEX | ANATOMY | GEOGRAPHY | USA STATISTICS | CHINA STATISTICS | RELIGION | JOBS |
 |
||
Lescol Capsules, Lescol XL Tablets (Reliant) | ||
|
- Drugs index - Manufacturers - Feedback 
|
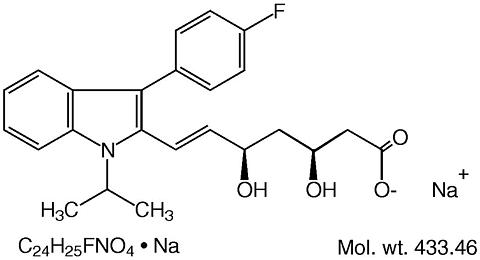
Prescribing Information DESCRIPTIONLescol ® (fluvastatin sodium), is a water-soluble cholesterol lowering agent which acts through the inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase. Fluvastatin sodium is [ R*, S *-( E )]-(±)-7-[3-(4-fluorophenyl)-1-(1-methylethyl)-1 H -indol-2-yl]-3,5-dihydroxy-6-heptenoic acid, monosodium salt. The empirical formula of fluvastatin sodium is C 24 H 25 FNO 4 ·Na, its molecular weight is 433.46 and its structural formula is:
This molecular entity is the first entirely synthetic HMG-CoA reductase inhibitor, and is in part structurally distinct from the fungal derivatives of this therapeutic class. Fluvastatin sodium is a white to pale yellow, hygroscopic powder soluble in water, ethanol and methanol. Lescol is supplied as capsules containing fluvastatin sodium, equivalent to 20 mg or 40 mg of fluvastatin, for oral administration. Lescol ® XL (fluvastatin sodium) is supplied as extended-release tablets containing fluvastatin sodium, equivalent to 80 mg of fluvastatin, for oral administration. Active Ingredient: fluvastatin sodium Inactive Ingredients in capsules: gelatin, magnesium stearate, microcrystalline cellulose, pregelatinized starch (corn), red iron oxide, sodium lauryl sulfate, talc, titanium dioxide, yellow iron oxide, and other ingredients. Capsules may also include: benzyl alcohol, black iron oxide, butylparaben, carboxymethylcellulose sodium, edetate calcium disodium, methylparaben, propylparaben, silicon dioxide and sodium propionate. Inactive Ingredients in extended-release tablets: microcrystalline cellulose, hydroxypropyl cellulose, hydroxypropyl methyl cellulose, potassium bicarbonate, povidone, magnesium stearate, iron oxide yellow, titanium dioxide and polyethylene glycol 8000.
CLINICAL PHARMACOLOGYA variety of clinical studies have demonstrated that elevated levels of total cholesterol (Total-C), low densitylipoprotein cholesterol (LDL-C), triglycerides (TG) andapolipoprotein B (a membrane transport complex for LDL-C) promote human atherosclerosis. Similarly, decreased levels of HDL-cholesterol (HDL-C) and its transport complex, apolipoprotein A, are associated with the development of atherosclerosis. Epidemiologic investigations have established that cardiovascular morbidity and mortality vary directly with the level of Total-C and LDL-C and inversely with the level of HDL-C. Like LDL, cholesterol-enriched triglyceride-rich lipoproteins, including VLDL, IDL and remnants, can also promote atherosclerosis. Elevated plasma triglycerides are frequently found in a triad with low HDL-C levels and small LDL particles, as well as in association with non-lipid metabolic risk factors for coronary heart disease. As such, total plasma TG has not consistently been shown to be an independent risk factor for CHD. Furthermore, the independent effect of raising HDL or lowering TG on the risk of coronary and cardiovascular morbidity and mortality has not been determined. In patients with hypercholesterolemia and mixed dyslipidemia, treatment with Lescol ® (fluvastatin sodium) or Lescol ® XL (fluvastatin sodium) reduced Total-C, LDL-C, apolipoprotein B, and triglycerides while producing an increase in HDL-C. Increases in HDL-C are greater in patients with low HDL-C (<35 mg/dL). Neither agent had a consistent effect on either Lp(a) or fibrinogen. The effect of Lescol or Lescol XL induced changes in lipoprotein levels, including reduction of serum cholesterol, on cardiovascular mortality has not been determined. Mechanism of ActionLescol is a competitive inhibitor of HMG-CoA reductase, which is responsible for the conversion of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) to mevalonate, a precursor of sterols, including cholesterol. The inhibition of cholesterol biosynthesis reduces the cholesterol in hepatic cells, which stimulates the synthesis of LDL receptors and thereby increases the uptake of LDL particles. The end result of these biochemical processes is a reduction of the plasma cholesterol concentration. Pharmacokinetics/MetabolismOral AbsorptionFluvastatin is absorbed rapidly and completely following oral administration of the capsule, with peak concentrations reached in less than 1 hour. Following administration of a 10 mg dose, the absolute bioavailability is 24% (range 9%-50%). Administration with food reduces the rate but not the extent of absorption. At steady-state, administration of fluvastatin with the evening meal results in a two-fold decrease in C max and more than two-fold increase in t max as compared to administration 4 hours after the evening meal. No significant differences in extent of absorption or in the lipid-lowering effects were observed between the two administrations. After single or multiple doses above 20 mg, fluvastatin exhibits saturable first-pass metabolism resulting in higher-than-expected plasma fluvastatin concentrations. Fluvastatin has two optical enantiomers, an active 3R,5S and an inactive 3S,5R form. In vivo studies showed that stereo-selective hepatic binding of the active form occurs during the first pass resulting in a difference in the peak levels of the two enantiomers, with the active to inactive peak concentration ratio being about 0.7. The approximate ratio of the active to inactive approaches unity after the peak is seen and thereafter the two enantiomers decline with the same half-life. After an intravenous administration, bypassing the first-pass metabolism, the ratios of the enantiomers in plasma were similar throughout the concentration-time profiles. Fluvastatin administered as Lescol XL 80 mg tablets reaches peak concentration in approximately 3 hours under fasting conditions, after a low-fat meal, or 2.5 hours after a low-fat meal. The mean relative bioavailability of the XL tablet is approximately 29% (range: 9%-66%) compared to that of the Lescol immediate release capsule administered under fasting conditions. Administration of a high fat meal delayed the absorption (T max : 6H) and increased the bioavailability of the XL tablet by approximately 50%. Once Lescol XL begins to be absorbed, fluvastatin concentrations rise rapidly. The maximum concentration seen after a high fat meal is much less than the peak concentration following a single dose or twice daily dose of the 40 mg Lescol capsule. Overall variability in the pharmacokinetics of Lescol XL is large (42%-64% CV for C max and AUC), and especially so after a high fat meal (63%-89% for C max and AUC). Intrasubject variability in the pharmacokinetics of Lescol XL under fasting conditions (about 25% for C max and AUC) tends to be much smaller as compared to the overall variability. Multiple peaks in plasma fluvastatin concentrations have been observed after Lescol XL administration. DistributionFluvastatin is 98% bound to plasma proteins. The mean volume of distribution (VD ss ) is estimated at 0.35 L/kg. The parent drug is targeted to the liver and no active metabolites are present systemically. At therapeutic concentrations, the protein binding of fluvastatin is not affected by warfarin, salicylic acid and glyburide. MetabolismFluvastatin is metabolized in the liver, primarily via hydroxylation of the indole ring at the 5- and 6-positions. N-dealkylation and beta-oxidation of the side-chain also occurs. The hydroxy metabolites have some pharmacologic activity, but do not circulate in the blood. Both enantiomers of fluvastatin are metabolized in a similar manner. In vitro studies demonstrated that fluvastatin undergoes oxidative metabolism, predominantly via 2C9 isozyme systems (75%). Other isozymes that contribute to fluvastatin metabolism are 2C8 (~5%) and 3A4 (~20%). (See PRECAUTIONS : Drug Interactions Section) . EliminationFluvastatin is primarily (about 90%) eliminated in the feces as metabolites, with less than 2% present as unchanged drug. Urinary recovery is about 5%. After a radiolabeled dose of fluvastatin, the clearance was 0.8 L/h/kg. Following multiple oral doses of radiolabeled compound, there was no accumulation of fluvastatin; however, there was a 2.3 fold accumulation of total radioactivity. Steady-state plasma concentrations show no evidence of accumulation of fluvastatin following immediate release capsule administration of up to 80 mg daily, as evidenced by a beta-elimination half-life of less than 3 hours. However, under conditions of maximum rate of absorption (i.e., fasting) systemic exposure to fluvastatin is increased 33% to 53% compared to a single 20 mg or 40 mg dose of the immediate release capsule. Following once daily administration of the 80 mg Lescol XL tablet for 7 days, systemic exposure to fluvastatin is increased (20%-30%) compared to a single dose of the 80 mg Lescol XL tablet. Terminal half-life of Lescol XL was about 9 hours as a result of the slow-release formulation. Single-dose and steady-state pharmacokinetic parameters in 33 subjects with hypercholesterolemia for the capsules and in 35 healthy subjects for the extended-release tablets are summarized below:
Special Populations
Renal Insufficiency: No significant (<6%) renal excretion of fluvastatin occurs in humans. Hepatic Insufficiency: Fluvastatin is subject to saturable first-pass metabolism/sequestration by the liver and is eliminated primarily via the biliary route. Therefore, the potential exists for drug accumulation in patients with hepatic insufficiency. Caution should therefore be exercised when fluvastatin sodium is administered to patients with a history of liver disease or heavy alcohol ingestion (see WARNINGS ). Fluvastatin AUC and C max values increased by about 2.5 fold in hepatic insufficiency patients. This result was attributed to the decreased presystemic metabolism due to hepatic dysfunction. The enantiomer ratios of the two isomers of fluvastatin in hepatic insufficiency patients were comparable to those observed in healthy subjects. Age: Plasma levels of fluvastatin are not affected by age. Gender: Women tend to have slightly higher (but statistically insignificant) fluvastatin concentrations than men for the immediate release capsule. This is most likely due to body weight differences, as adjusting for body weight decreases the magnitude of the differences seen. For Lescol XL, there are 67% and 77% increases in systemic availability for women over men under fasted and high fat meal conditions. Pediatric: No data are available. Fluvastatin is not indicated for use in the pediatric population.
CLINICAL STUDIESHypercholesterolemia (heterozygous familial and non familial) and Mixed DyslipidemiaIn 12 placebo-controlled studies in patients with Type IIa or IIb hyperlipoproteinemia, Lescol ® (fluvastatin sodium) alone was administered to 1621 patients in daily dose regimens of 20 mg, 40 mg, and 80 mg (40 mg twice daily) for at least 6 weeks duration. After 24 weeks of treatment, daily doses of 20 mg, 40 mg, and 80 mg (40 mg twice daily) resulted in median LDL-C reductions of 22% (n=747), 25% (n=748) and 36% (n=257), respectively. Lescol treatment produced dose-related reductions in Apo B and in triglycerides and increases in HDL-C. The median (25 th , 75 th percentile) percent changes from baseline in HDL-C after 12 weeks of treatment with Lescol at daily doses of 20 mg, 40 mg and 80 mg (40 mg twice daily) were +2 (-4,+10), +5 (-2,+12), and +4 (-3,+12), respectively. In a subgroup of patients with primary mixed dyslipidemia, defined as baseline TG levels >/=200 mg/dL, treatment with Lescol also produced significant decreases in Total-C, LDL-C, TG and Apo B and variable increases in HDL-C. The median (25 th 75 th percentile) percent changes from baseline in HDL-C after 12 weeks of treatment with Lescol at daily doses of 20 mg, 40 mg and 80 mg (40 mg twice daily) in this population were +4 (-2,+12), +8 (+1,+15), and +4 (-3,+13), respectively. In a long-term open-label free titration study, after 96 weeks LDL-C decreases of 25% (20 mg, n=68), 31% (40 mg, n=298) and 34% (80 mg, n=209) were seen. No consistent effect on Lp(a) was observed. Lescol ® XL (fluvastatin sodium) Extended-Release Tablets have been studied in five controlled studies of patients with Type IIa or IIb hyperlipoproteinemia. Lescol XL was administered to over 900 patients in trials from 4 to 26 weeks in duration. In the three largest of these studies, Lescol XL given as a single daily dose of 80 mg significantly reduced Total-C, LDL-C, TG and Apo B. Therapeutic response is well established within two weeks, and a maximum response is achieved within four weeks. After four weeks of therapy, the median decrease in LDL-C was 38% and at week 24 endpoint the median LDL-C decrease was 35%. Significant increases in HDL-C were also observed. The median (25 th and 75 th percentile) percent changes from baseline in HDL-C for Lescol XL were +7(+0,+15) after 24 weeks of treatment.
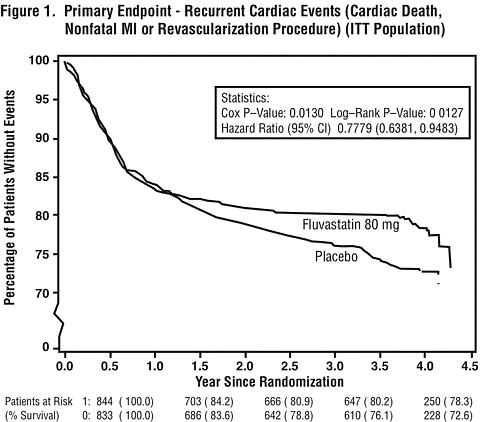
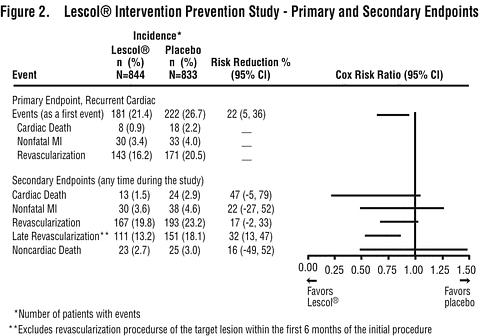
In patients with primary mixed dyslipidemia (Fredrickson Type IIb) as defined by baseline plasma triglycerides levels >/=200 mg/dL, Lescol XL 80 mg produced a median reduction in triglycerides of 25%. In these patients, Lescol XL 80 mg produced median (25 th and 75 th percentile) percent change from baseline in HDL-C of +11(+3,+20). Significant decreases in Total-C, LDL-C, and Apo B were also achieved. In these studies, patients with triglycerides >400 mg/dL were excluded. Reduction in the Risk of Recurrent Cardiac EventsIn the Lescol Intervention Prevention Study, the effect of Lescol 40 mg administered twice daily on the risk of recurrent cardiac events (time to first occurrence of cardiac death, nonfatal myocardial infarction, or revascularization) was assessed in 1677 patients with coronary heart disease who had undergone a percutaneous coronary intervention (PCI) procedure (mean time from PCI to randomization=3 days). In this multicenter, randomized, double-blind, placebo-controlled study, patients were treated with dietary/lifestyle counseling and either Lescol 40 mg (n=844) or placebo (n=833) given twice daily for a median of 3.9 years. The study population was 84% male, 98% Caucasian, with 37% >65 years of age. At baseline patients had total cholesterol between 100 and 367 mg/dL (mean 201 mg/dL), LDL-C between 42 and 243 mg/dL (mean 132 mg/dL), triglycerides between 15 and 270 mg/dL (mean 70 mg/dL) and HDL-C between 8 and 174 mg/dL (mean 39 mg/dL). Lescol significantly reduced the risk of recurrent cardiac events (Figure 1) by 22% (p=0.013, 181 patients in the Lescol group vs. 222 patients in the placebo group). Revascularization procedures comprised the majority of the initial recurrent cardiac events (143 revascularization procedures in the Lescol group and 171 in the placebo group). Consistent trends in risk reduction were observed in patients >65 years of age.
Outcome data for the Lescol Intervention Prevention Study are shown in Figure 2. After exclusion of revascularization procedures (CABG and repeat PCI) occurring within the first 6 months of the initial procedure involving the originally instrumented site, treatment with Lescol was associated with a 32% (p=0.002) reduction in risk of late revascularization procedures (CABG or PCI occurring at the original site >6 months after the initial procedure, or at another site).
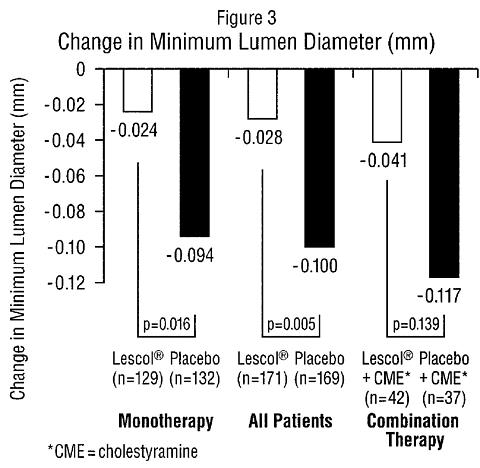
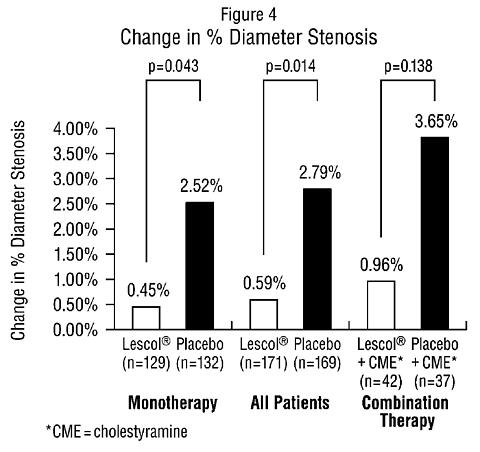
AtherosclerosisIn the Lipoprotein and Coronary Atherosclerosis Study (LCAS), the effect of Lescol therapy on coronary atherosclerosis was assessed by quantitative coronary angiography (QCA) in patients with coronary artery disease and mild to moderate hypercholesterolemia (baseline LDL-C range 115-190 mg/dL). In this randomized double-blind, placebo controlled trial, 429 patients were treated with conventional measures (Step 1 AHA Diet) and either Lescol 40 mg/day or placebo. In order to provide treatment to patients receiving placebo with LDL-C levels >/=160 mg/dL at baseline, adjunctive therapy with cholestyramine was added after week 12 to all patients in the study with baseline LDL-C values of >/=160 mg/dL. These baseline levels were present in 25% of the study population. Quantitative coronary angiograms were evaluated at baseline and 2.5 years in 340 (79%) angiographic evaluable patients. Lescol significantly slowed the progression of coronary atherosclerosis. Compared to placebo, Lescol significantly slowed the progression of lesions as measured by within-patient per-lesion change in minimum lumen diameter (MLD), the primary endpoint (see Figure 3 below), percent diameter stenosis (Figure 4), and the formation of new lesions (13% of all fluvastatin patients versus 22% of all placebo patients). Additionally, a significant difference in favor of Lescol was found between all fluvastatin and all placebo patients in the distribution among the three categories of definite progression, definite regression, and mixed or no change. Beneficial angiographic results (change in MLD) were independent of patients' gender and consistent across a range of baseline LDL-C levels.
INDICATIONS AND USAGETherapy with lipid-altering agents should be used in addition to a diet restricted in saturated fat and cholesterol (see National Cholesterol Education Program (NCEP) Treatment Guidelines, below). Hypercholesterolemia (heterozygous familial and non familial) and Mixed DyslipidemiaLescol ® (fluvastatin sodium) and Lescol ® XL (fluvastatin sodium) are indicated to reduce elevated total cholesterol (Total-C), LDL-C, TG and Apo B levels, and to increase HDL-C in patients with primary hypercholesterolemia and mixed dyslipidemia (Fredrickson Type IIa and IIb) whose response to dietary restriction of saturated fat and cholesterol and other nonpharmacological measures has not been adequate. Secondary Prevention of Coronary EventsIn patients with coronary heart disease, Lescol and Lescol XL are indicated to reduce the risk of undergoing coronary revascularization procedures. Atherosclerosis Lescol and Lescol XL are also indicated to slow the progression of coronary atherosclerosis in patients with coronary heart disease as part of a treatment strategy to lower total and LDL cholesterol to target levels. Therapy with lipid-altering agents should be considered only after secondary causes for hyperlipidemia such as poorly controlled diabetes mellitus, hypothyroidism, nephrotic syndrome, dysproteinemias, obstructive liver disease, other medication, or alcoholism, have been excluded. Prior to initiation of fluvastatin sodium, a lipid profile should be performed to measure Total-C, HDL-C and TG. For patients with TG <400 mg/dL (<4.5 mmol/L), LDL-C can be estimated using the following equation: LDL-C = Total-C - HDL-C - 1/5 TG For TG levels >400 mg/dL (>4.5 mmol/L), this equation is less accurate and LDL-C concentrations should be determined by ultracentrifugation. In many hypertriglyceridemic patients LDL-C may be low or normal despite elevated Total-C. In such cases, Lescol is not indicated. Lipid determinations should be performed at intervals of no less than 4 weeks and dosage adjusted according to the patient's response to therapy. The National Cholesterol Education Program (NCEP) Treatment Guidelines are summarized below:
After the LDL-C goal has been achieved, if the TG is still >/=200 mg/dL, non-HDL-C (total-C minus HDL-C) becomes a secondary target of therapy. Non-HDL-C goals are set 30 mg/dL higher than LDL-C goals for each risk category. At the time of hospitalization for an acute coronary event, consideration can be given to initiating drug therapy at discharge if the LDL-C level is >/=130 mg/dL (NCEP-ATP II). Since the goal of treatment is to lower LDL-C, the NCEP recommends that the LDL-C levels be used to initiate and assess treatment response. Only if LDL-C levels are not available, should the Total-C be used to monitor therapy.
Neither Lescol nor Lescol XL have been studied in conditions where the major abnormality is elevation of chylomicrons, VLDL, or IDL (i.e., hyperlipoproteinemia Types I, III, IV, or V).
CONTRAINDICATIONSHypersensitivity to any component of this medication. Lescol ® (fluvastatin sodium) and Lescol ® XL (fluvastatin sodium) are contraindicated in patients with active liver disease or unexplained, persistent elevations in serum transaminases (see WARNINGS). Pregnancy and LactationAtherosclerosis is a chronic process and discontinuation of lipid-lowering drugs during pregnancy should have little impact on the outcome of long-term therapy of primary hypercholesterolemia. Cholesterol and other products of cholesterol biosynthesis are essential components for fetal development (including synthesis of steroids and cell membranes). Since HMG-CoA reductase inhibitors decrease cholesterol synthesis and possibly the synthesis of other biologically active substances derived from cholesterol, they may cause fetal harm when administered to pregnant women. Therefore, HMG-CoA reductase inhibitors are contraindicated during pregnancy and in nursing mothers. Fluvastatin sodium should be administered to women of childbearing age only when such patients are highly unlikely to conceive and have been informed of the potential hazards. If the patient becomes pregnant while taking this class of drug, therapy should be discontinued and the patient apprised of the potential hazard to the fetus.
WARNINGSLiver EnzymesBiochemical abnormalities of liver function have been associated with HMG-CoA reductase inhibitors and other lipid-lowering agents. Approximately 1.1% of patients treated with Lescol ® (fluvastatin sodium) capsules in worldwide trials developed dose-related, persistent elevations of transaminase levels to more than 3 times the upper limit of normal. Fourteen of these patients (0.6%) were discontinued from therapy. In all clinical trials, a total of 33/2969 patients (1.1%) had persistent transaminase elevations with an average fluvastatin exposure of approximately 71.2 weeks; 19 of these patients (0.6%) were discontinued. The majority of patients with these abnormal biochemical findings were asymptomatic. In a pooled analysis of all placebo-controlled studies in which Lescol capsules were used, persistent transaminase elevations (>3 times the upper limit of normal [ULN] on two consecutive weekly measurements) occurred in 0.2%, 1.5%, and 2.7% of patients treated with 20, 40, and 80 mg (titrated to 40 mg twice daily) Lescol capsules, respectively. Ninety-one percent of the cases of persistent liver function test abnormalities (20 of 22 patients) occurred within 12 weeks of therapy and in all patients with persistent liver function test abnormalities there was an abnormal liver function test present at baseline or by week 8. In the pooled analysis of the 24-week controlled trials, persistent transaminase elevation occurred in 1.9%, 1.8% and 4.9% of patients treated with Lescol ® XL (fluvastatin sodium) 80 mg, Lescol 40 mg and Lescol 40 mg twice daily, respectively. In 13 of 16 patients treated with Lescol XL the abnormality occurred within 12 weeks of initiation of treatment with Lescol XL 80 mg. It is recommended that liver function tests be performed before the initiation of therapy and at 12 weeks following initiation of treatment or elevation in dose. Patients who develop transaminase elevations or signs and symptoms of liver disease should be monitored to confirm the finding and should be followed thereafter with frequent liver function tests until the levels return to normal. Should an increase in AST or ALT of three times the upper limit of normal or greater persist (found on two consecutive occasions) withdrawal of fluvastatin sodium therapy is recommended. Active liver disease or unexplained transaminase elevations are contraindications to the use of Lescol and Lescol XL (see CONTRAINDICATIONS ) . Caution should be exercised when fluvastatin sodium is administered to patients with a history of liver disease or heavy alcohol ingestion (see CLINICAL PHARMACOLOGY : Pharmacokinetics/Metabolism ) . Such patients should be closely monitored. Skeletal MuscleRhabdomyolysis with renal dysfunction secondary to myoglobinuria has been reported with fluvastatin and with other drugs in this class. Myopathy, defined as muscle aching or muscle weakness in conjunction with increases in creatine phosphokinase (CPK) values to greater than 10 times the upper limit of normal, has been reported. Myopathy should be considered in any patients with diffuse myalgias, muscle tenderness or weakness, and/or marked elevation of CPK. Patients should be advised to report promptly unexplained muscle pain, tenderness or weakness, particularly if accompanied by malaise or fever. Fluvastatin sodium therapy should be discontinued if markedly elevated CPK levels occur or myopathy is diagnosed or suspected. Fluvastatin sodium therapy should also be temporarily withheld in any patient experiencing an acute or serious condition predisposing to the development of renal failure secondary to rhabdomyolysis, e.g., sepsis; hypotension; major surgery; trauma; severe metabolic, endocrine, or electrolyte disorders; or uncontrolled epilepsy. The risk of myopathy and or rhabdomyolysis during treatment with HMG-CoA reductase inhibitors has been reported to be increased if therapy with either cyclosporine, gemfibrozil, erythromycin, or niacin is administered concurrently. Myopathy was not observed in a clinical trial in 74 patients involving patients who were treated with fluvastatin sodium together with niacin. Uncomplicated myalgia has been observed infrequently in patients treated with Lescol at rates indistinguishable from placebo. The use of fibrates alone may occasionally be associated with myopathy. The combined use of HMG-CoA reductase inhibitors and fibrates should generally be avoided.
PRECAUTIONSGeneralBefore instituting therapy with Lescol ® (fluvastatin sodium) or Lescol ® XL (fluvastatin sodium), an attempt should be made to control hypercholesterolemia with appropriate diet, exercise, and weight reduction in obese patients, and to treat other underlying medical problems (see INDICATIONS AND USAGE ) . The HMG-CoA reductase inhibitors may cause elevation of creatine phosphokinase and transaminase levels (see WARNINGS and ADVERSE REACTIONS ) . This should be considered in the differential diagnosis of chest pain in a patient on therapy with fluvastatin sodium. Homozygous Familial HypercholesterolemiaHMG-CoA reductase inhibitors are reported to be less effective in patients with rare homozygous familial hypercholesterolemia, possibly because these patients have few functional LDL receptors. Information for PatientsPatients should be advised to report promptly unexplained muscle pain, tenderness or weakness, particularly if accompanied by malaise or fever. Women should be informed that if they become pregnant while receiving Lescol or Lescol XL the drug should be discontinued immediately to avoid possible harmful effects on a developing fetus from a relative deficit of cholesterol and biological products derived from cholesterol. In addition, Lescol or Lescol XL should not be taken during nursing. (See CONTRAINDICATIONS .) Drug InteractionsThe below listed drug interaction information is derived from studies using immediate release fluvastatin. Similar studies have not been conducted using the Lescol XL tablet. Immunosuppressive Drugs, Gemfibrozil, Niacin (Nicotinic Acid), Erythromycin (See WARNINGS : Skeletal Muscle ) . In vitro data indicate that fluvastatin metabolism involves multiple Cytochrome P450 (CYP) isozymes. CYP2C9 isoenzyme is primarily involved in the metabolism of fluvastatin (~75%), while CYP2C8 and CYP3A4 isoenzymes are involved to a much less extent, i.e. ~5% and ~20%, respectively. If one pathway is inhibited in the elimination process of fluvastatin other pathways may compensate. In vivo drug interaction studies with CYP3A4 inhibitors/substrates such as cyclosporine, erythromycin, and itraconazle result in minimal changes in the pharmacokinetics of fluvastatin, confirming less involvement of CYP3A4 isozyme. Concomitant administration of fluvastatin and phenytoin increased the levels of phenytoin and fluvastatin, suggesting predominant involvement of CYP2C9 in fluvastatin metabolism. Niacin/Propranolol: Concomitant administration of immediate release fluvastatin sodium with niacin or propranolol has no effect on the bioavailability of fluvastatin sodium. Cholestyramine: Administration of immediate release fluvastatin sodium concomitantly with, or up to 4 hours after cholestyramine, results in fluvastatin decreases of more than 50% for AUC and 50%-80% for C max . However, administration of immediate release fluvastatin sodium 4 hours after cholestyramine resulted in a clinically significant additive effect compared with that achieved with either component drug. Cyclosporine: Plasma cyclosporine levels remain unchanged when fluvastatin (20 mg daily) was administered concurrently in renal transplant recipients on stable cyclosporine regimens. Fluvastatin AUC increased 1.9 fold, and C max increased 1.3 fold compared to historical controls. Digoxin: In a crossover study involving 18 patients chronically receiving digoxin, a single 40 mg dose of immediate release fluvastatin had no effect on digoxin AUC, but had an 11% increase in digoxin C max and small increase in digoxin urinary clearance. Erythromycin: Erythromycin (500 mg, single dose) did not affect steady-state plasma levels of fluvastatin (40 mg daily). Itraconazole: Concomitant administration of fluvastatin (40 mg) and itraconazole (100 mg daily × 4 days) does not affect plasma itraconazole or fluvastatin levels. Gemfibrozil: There is no change in either fluvastatin (20 mg twice daily) or gemfibrozil (600 mg twice daily) plasma levels when these drugs are co-administered. Phenytoin: Single morning dose administration of phenytoin (300 mg extended release) increased mean steady-state fluvastatin (40 mg) C max by 27% and AUC by 40% whereas fluvastatin increased the mean phenytoin C max by 5% and AUC by 20%. Patients on phenytoin should continue to be monitored appropriately when fluvastatin therapy is initiated or when the fluvastatin dosage is changed. Diclofenac: Concurrent administration of fluvastatin (40 mg) increased the mean C max and AUC of diclofenac by 60% and 25% respectively. Tolbutamide: In healthy volunteers, concurrent administration of either single or multiple daily doses of fluvastatin sodium (40 mg) with tolbutamide (1 g) did not affect the plasma levels of either drug to a clinically significant extent. Glibenclamide (Glyburide): In glibenclamide-treated NIDDM patients (n=32), administration of fluvastatin (40 mg twice daily for 14 days) increased the mean C max , AUC, and t 1/2 of glibenclamide approximately 50%, 69% and 121%, respectively. Glibenclamide (5-20 mg daily) increased the mean C max and AUC of fluvastatin by 44% and 51%, respectively. In this study there were no changes in glucose, insulin and C-peptide levels. However, patients on concomitant therapy with glibenclamide (glyburide) and fluvastatin should continue to be monitored appropriately when their fluvastatin dose is increased to 40 mg twice daily. Losartan: Concomitant administration of fluvastatin with losartan has no effect on the bioavailability of either losartan or its active metabolite. Cimetidine/Ranitidine/Omeprazole: Concomitant administration of immediate release fluvastatin sodium with cimetidine, ranitidine and omeprazole results in a significant increase in the fluvastatin C max (43%, 70% and 50%, respectively) and AUC (24%-33%), with an 18%-23% decrease in plasma clearance. Rifampicin: Administration of immediate release fluvastatin sodium to subjects pretreated with rifampicin results in significant reduction in C max (59%) and AUC (51%), with a large increase (95%) in plasma clearance. Warfarin: In vitro protein binding studies demonstrated no interaction at therapeutic concentrations. Concomitant administration of a single dose of warfarin (30 mg) in young healthy males receiving immediate release fluvastatin sodium (40 mg/day × 8 days) resulted in no elevation of racemic warfarin concentration. There was also no effect on prothrombin complex activity when compared to concomitant administration of placebo and warfarin. However, bleeding and/or increased prothrombin times have been reported in patients taking coumarin anticoagulants concomitantly with other HMG-CoA reductase inhibitors. Therefore, patients receiving warfarin-type anticoagulants should have their prothrombin times closely monitored when fluvastatin sodium is initiated or the dosage of fluvastatin sodium is changed. Endocrine FunctionHMG-CoA reductase inhibitors interfere with cholesterol synthesis and lower circulating cholesterol levels and, as such, might theoretically blunt adrenal or gonadal steroid hormone production. Fluvastatin exhibited no effect upon non-stimulated cortisol levels and demonstrated no effect upon thyroid metabolism as assessed by TSH. Small declines in total testosterone have been noted in treated groups, but no commensurate elevation in LH occurred, suggesting that the observation was not due to a direct effect upon testosterone production. No effect upon FSH in males was noted. Due to the limited number of premenopausal females studied to date, no conclusions regarding the effect of fluvastatin upon female sex hormones may be made. Two clinical studies in patients receiving fluvastatin at doses up to 80 mg daily for periods of 24 to 28 weeks demonstrated no effect of treatment upon the adrenal response to ACTH stimulation. A clinical study evaluated the effect of fluvastatin at doses up to 80 mg daily for 28 weeks upon the gonadal response to HCG stimulation. Although the mean total testosterone response was significantly reduced (p<0.05) relative to baseline in the 80 mg group, it was not significant in comparison to the changes noted in groups receiving either 40 mg of fluvastatin or placebo. Patients treated with fluvastatin sodium who develop clinical evidence of endocrine dysfunction should be evaluated appropriately. Caution should be exercised if an HMG-CoA reductase inhibitor or other agent used to lower cholesterol levels is administered to patients receiving other drugs (e.g., ketoconazole, spironolactone, or cimetidine) that may decrease the levels of endogenous steroid hormones. CNS ToxicityCNS effects, as evidenced by decreased activity, ataxia, loss of righting reflex, and ptosis were seen in the following animal studies: the 18-month mouse carcinogenicity study at 50 mg/kg/day, the 6-month dog study at 36 mg/kg/day, the 6-month hamster study at 40 mg/kg/day, and in acute, high-dose studies in rats and hamsters (50 mg/kg), rabbits (300 mg/kg) and mice (1500 mg/kg). CNS toxicity in the acute high-dose studies was characterized (in mice) by conspicuous vacuolation in the ventral white columns of the spinal cord at a dose of 5000 mg/kg and (in rat) by edema with separation of myelinated fibers of the ventral spinal tracts and sciatic nerve at a dose of 1500 mg/kg. CNS toxicity, characterized by periaxonal vacuolation, was observed in the medulla of dogs that died after treatment for 5 weeks with 48 mg/kg/day; this finding was not observed in the remaining dogs when the dose level was lowered to 36 mg/kg/day. CNS vascular lesions, characterized by perivascular hemorrhages, edema, and mononuclear cell infiltration of perivascular spaces, have been observed in dogs treated with other members of this class. No CNS lesions have been observed after chronic treatment for up to 2 years with fluvastatin in the mouse (at doses up to 350 mg/kg/day), rat (up to 24 mg/kg/day), or dog (up to 16 mg/kg/day). Prominent bilateral posterior Y suture lines in the ocular lens were seen in dogs after treatment with 1, 8, and 16 mg/kg/day for 2 years. Carcinogenesis, Mutagenesis, Impairment of FertilityA 2-year study was performed in rats at dose levels of 6, 9, and 18-24 (escalated after 1 year) mg/kg/day. These treatment levels represented plasma drug levels of approximately 9, 13, and 26-35 times the mean human plasma drug concentration after a 40 mg oral dose. A low incidence of forestomach squamous papillomas and 1 carcinoma of the forestomach at the 24 mg/kg/day dose level was considered to reflect the prolonged hyperplasia induced by direct contact exposure to fluvastatin sodium rather than to a systemic effect of the drug. In addition, an increased incidence of thyroid follicular cell adenomas and carcinomas was recorded for males treated with 18-24 mg/kg/day. The increased incidence of thyroid follicular cell neoplasm in male rats with fluvastatin sodium appears to be consistent with findings from other HMG-CoA reductase inhibitors. In contrast to other HMG-CoA reductase inhibitors, no hepatic adenomas or carcinomas were observed. The carcinogenicity study conducted in mice at dose levels of 0.3, 15 and 30 mg/kg/day revealed, as in rats, a statistically significant increase in forestomach squamous cell papillomas in males and females at 30 mg/kg/day and in females at 15 mg/kg/day. These treatment levels represented plasma drug levels of approximately 0.05, 2, and 7 times the mean human plasma drug concentration after a 40 mg oral dose. No evidence of mutagenicity was observed in vitro, with or without rat-liver metabolic activation, in the following studies: microbial mutagen tests using mutant strains of Salmonella typhimurium or Escherichia coli; malignant transformation assay in BALB/3T3 cells; unscheduled DNA synthesis in rat primary hepatocytes; chromosomal aberrations in V79 Chinese Hamster cells; HGPRT V79 Chinese Hamster cells. In addition, there was no evidence of mutagenicity in vivo in either a rat or mouse micronucleus test. In a study in rats at dose levels for females of 0.6, 2 and 6 mg/kg/day and at dose levels for males of 2, 10 and 20 mg/kg/day, fluvastatin sodium had no adverse effects on the fertility or reproductive performance. Seminal vesicles and testes were small in hamsters treated for 3 months at 20 mg/kg/day (approximately three times the 40 milligram human daily dose based on surface area, mg/m 2 ). There was tubular degeneration and aspermatogenesis in testes as well as vesiculitis of seminal vesicles. Vesiculitis of seminal vesicles and edema of the testes were also seen in rats treated for 2 years at 18 mg/kg/day (approximately 4 times the human C max achieved with a 40 milligram daily dose).
Pregnancy
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The following effects have been reported with drugs in this class. Not all the effects listed below have necessarily been associated with fluvastatin sodium therapy.
Skeletal: muscle cramps, myalgia, myopathy, rhabdomyolysis, arthralgias.
Neurological: dysfunction of certain cranial nerves (including alteration of taste, impairment of extra-ocular movement, facial paresis), tremor, dizziness, vertigo, memory loss, paresthesia, peripheral neuropathy, peripheral nerve palsy, psychic disturbances, anxiety, insomnia, depression.
Hypersensitivity Reactions: An apparent hypersensitivity syndrome has been reported rarely which has included one or more of the following features: anaphylaxis, angioedema, lupus erythematosus-like syndrome, polymyalgia rheumatica, vasculitis, purpura, thrombocytopenia, leukopenia, hemolytic anemia, positive ANA, ESR increase, eosinophilia, arthritis, arthralgia, urticaria, asthenia, photosensitivity, fever, chills, flushing, malaise, dyspnea, toxic epidermal necrolysis, erythema multiforme, including Stevens-Johnson syndrome.
Gastrointestinal: pancreatitis, hepatitis, including chronic active hepatitis, cholestatic jaundice, fatty change in liver, and, rarely, cirrhosis, fulminant hepatic necrosis, and hepatoma; anorexia, vomiting.
Skin: alopecia, pruritus. A variety of skin changes (e.g., nodules, discoloration, dryness of skin/mucous membranes, changes to hair/nails) have been reported.
Reproductive: gynecomastia, loss of libido, erectile dysfunction.
Eye: progression of cataracts (lens opacities), ophthalmoplegia.
Laboratory Abnormalities: elevated transaminases, alkaline phosphatase, (gamma)-glutamyl transpeptidase, and bilirubin; thyroid function abnormalities.
Fluvastatin sodium has been administered concurrently with cholestyramine and nicotinic acid. No adverse reactions unique to the combination or in addition to those previously reported for this class of drugs alone have been reported. Myopathy and rhabdomyolysis (with or without acute renal failure) have been reported when another HMG-CoA reductase inhibitor was used in combination with immunosuppressive drugs, gemfibrozil, erythromycin, or lipid-lowering doses of nicotinic acid. Concomitant therapy with HMG-CoA reductase inhibitors and these agents is generally not recommended. (See WARNINGS : Skeletal Muscle .)
The approximate oral LD 50 is greater than 2 g/kg in mice and greater than 0.7 g/kg in rats.
The maximum single oral dose of Lescol ® (fluvastatin sodium) capsules received by healthy volunteers was 80 mg. No clinically significant adverse experiences were seen at this dose. The maximum dose administered with an extended-release formulation was 640 mg for two weeks. This dose was not well tolerated and produced a variety of GI complaints and an increase in transaminase values (i.e., SGOT and SGPT).
There has been a single report of 2 children, one 2 years old and the other 3 years of age, either of whom may have possibly ingested fluvastatin sodium. The maximum amount of fluvastatin sodium that could have been ingested was 80 mg (4 × 20 mg capsules). Vomiting was induced by ipecac in both children and no capsules were noted in their emesis. Neither child experienced any adverse symptoms and both recovered from the incident without problems.
Should an accidental overdose occur, treat symptomatically and institute supportive measures as required. The dialyzability of fluvastatin sodium and of its metabolites in humans is not known at present.
Information about the treatment of overdose can often be obtained from a certified Regional Poison Control Center. Telephone numbers of certified Regional Poison Control Centers are listed in the Physicians' Desk Reference ® . *
The patient should be placed on a standard cholesterol-lowering diet before receiving Lescol ® (fluvastatin sodium) or Lescol ® XL (fluvastatin sodium) and should continue on this diet during treatment with Lescol or Lescol XL. (See NCEP Treatment Guidelines for details on dietary therapy.)
For patients requiring LDL-C reduction to a goal of >/=25%, the recommended starting dose is 40 mg as one capsule, 80 mg as one Lescol XL tablet administered as a single dose in the evening or 80 mg in divided doses of the 40 mg capsule given twice daily. For patients requiring LDL-C reduction to a goal of <25% a starting dose of 20 mg may be used. The recommended dosing range is 20-80 mg/day. Lescol or Lescol XL may be taken without regard to meals, since there are no apparent differences in the lipid-lowering effects of fluvastatin sodium administered with the evening meal or 4 hours after the evening meal. Since the maximal reductions in LDL-C of a given dose are seen within 4 weeks, periodic lipid determinations should be performed and dosage adjustment made according to the patient's response to therapy and established treatment guidelines. The therapeutic effect of Lescol or Lescol XL is maintained with prolonged administration.
Lipid-lowering effects on total cholesterol and LDL cholesterol are additive when immediate release Lescol is combined with a bile-acid binding resin or niacin. When administering a bile-acid resin (e.g., cholestyramine) and fluvastatin sodium, Lescol should be administered at bedtime, at least 2 hours following the resin to avoid a significant interaction due to drug binding to resin. (See also ADVERSE REACTIONS : Concomitant Therapy .)
Since fluvastatin sodium is cleared hepatically with less than 6% of the administered dose excreted into the urine, dose adjustments for mild to moderate renal impairment are not necessary. Fluvastatin has not been studied at doses greater than 40 mg in patients with severe renal impairment; therefore caution should be exercised when treating such patients at higher doses.
Lescol ® (fluvastatin sodium) Capsules
20 mg
Brown and light brown imprinted twice with
|
|
Bottles of 30 capsules(NDC 0078-0176-15)
Bottles of 100 capsules(NDC 0078-0176-05)
40 mg
Brown and gold imprinted twice with
|
|
Bottles of 30 capsules(NDC 0078-0234-15)
Bottles of 100 capsules(NDC 0078-0234-05)
Lescol ® XL (fluvastatin sodium) Extended-Release Tablets
80 mg
Yellow, round, slightly biconvex film-coated tablet with beveled edges debossed with "Lescol XL" on one side and "80" on the other.
Bottles of 30 tablets(NDC 0078-0354-15)
Bottle of 100 tablets(NDC 0078-0354-05)
Store and Dispense
Store at 25°C (77°F); excursions permitted to 15°C-30°C (59°F-86°F). [See USP Controlled Room Temperature]. Dispense in a tight container. Protect from light.
T2003-40
REV: MAY 2003 89011106
 - To bookmark this page (add it to your favorites), please click the image to the left.
- To bookmark this page (add it to your favorites), please click the image to the left.